VANCOUVER, British Columbia–(BUSINESS WIRE)– Zymeworks Inc. (NYSE/TSX: ZYME), a clinical-stage biopharmaceutical company developing multifunctional biotherapeutics, today announced the appointment of two new independent members to its Board of Directors, further enhancing the Board’s commercial expertise. The new board members are Sue Mahony, Ph.D., MBA, a pharmaceutical executive with over 30 years of combined experience with Eli Lilly and Company (Lilly), Schering-Plough, Amgen, and Bristol-Myers Squibb, and Troy Cox, MBA, who has served in numerous senior leadership positions at leading biopharmaceutical companies such as Foundation Medicine, Roche-Genentech, UCB BioPharma, Sanofi-Aventis, and Schering-Plough.
“Sue Mahony and Troy Cox are highly respected biopharmaceutical executives that will provide essential leadership as Zymeworks transitions into a late-stage biotechnology company,” said Ali Tehrani, Ph.D., President and CEO of Zymeworks. “Both bring a wealth of knowledge and expertise in the strategic development and commercialization of therapeutics globally. We look forward to benefiting from their guidance as we continue to advance our robust clinical pipeline towards commercialization.”
SOURCE: Forté Foundation | Sue Mahony (above)
Sue Mahony brings extensive operational, clinical, and commercialization experience. Most recently, she served as Senior Vice President of Lilly and President of Lilly Oncology. While at Lilly, she held numerous senior leadership positions and led organizations in Europe, the US, Canada, Japan, and China; successfully developed, launched, and commercialized several global brands; and was a member of Lilly’s Executive Committee for 10 years. Prior to joining Lilly, Dr. Mahony served in sales and marketing roles in Europe for over a decade for Schering-Plough, Amgen, and Bristol-Myers Squibb. She also serves on the Board of Directors of Assembly Biosciences and Vifor Pharma. Dr. Mahony received a B.Sc. and a Ph.D. from Aston University and an MBA from London Business School.
SOURCE: Massachusetts Biotechnology Council | Troy Cox (above)
Troy Cox brings extensive biotech business and executive leadership experience to Zymeworks’ Board of Directors. Formerly, he served as President, Chief Executive Officer, and on the Board of Directors of Foundation Medicine, Inc., where he helped to advance oncology molecular science, partnered with biopharma, and secured reimbursement to deliver more advanced oncology care, thus shaping the oncology research, diagnostic, and data industries. Mr. Cox has also served as Senior Vice President, Sales & Marketing at Genentech, leading the oncology portfolio, as President of Central Nervous System (CNS) Operations at UCB BioPharma with responsibility for developing and commercializing therapeutics for complex CNS diseases, and in various senior commercial leadership roles at Sanofi-Aventis and Schering-Plough. Mr. Cox received a B.B.A. in finance from the University of Kentucky and an MBA from the University of Missouri.
About Zymeworks Inc.
Zymeworks is a clinical-stage biopharmaceutical company dedicated to the development of next-generation multifunctional biotherapeutics. The Company’s suite of therapeutic platforms and its fully integrated drug development engine enable precise engineering of highly differentiated product candidates. Zymeworks’ lead clinical candidate, ZW25, is a novel Azymetric™ bispecific antibody currently in Phase 2 clinical development. The Company’s second clinical candidate, ZW49, is a bispecific antibody-drug conjugate currently in Phase 1 clinical development and combines the unique design and antibody framework of ZW25 with Zymeworks’ proprietary ZymeLink™ cytotoxic payload. Zymeworks is also advancing a deep preclinical pipeline in immuno-oncology and other therapeutic areas. In addition, its therapeutic platforms are being leveraged through strategic partnerships with nine biopharmaceutical companies.
This press release includes “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. Forward-looking statements in this news release include, but are not limited to, statements that relate to the expected leadership to be provided by the new directors, Zymeworks’ anticipated advancement of its clinical pipeline towards commercialization and of its preclinical pipeline, advancements in Zymeworks’ partners therapeutic programs, the speed and outcome of drug development plans, and other information that is not historical information. When used herein, words such as “enable”, “plan”, “expect”, “will”, “may”, “look forward to”, “continue” and similar expressions are intended to identify forward-looking statements. In addition, any statements or information that refer to expectations, beliefs, plans, projections, objectives, performance or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking. All forward-looking statements are based upon Zymeworks’ current expectations and various assumptions. Zymeworks believes there is a reasonable basis for its expectations and beliefs, but they are inherently uncertain. Zymeworks may not realize its expectations, and its beliefs may not prove correct. Actual results could differ materially from those described or implied by such forward-looking statements as a result of various factors, including, without limitation, market conditions and the factors described under “Risk Factors” in Zymeworks’ Quarterly Report on Form 10-Q for its fiscal quarter ended March 31, 2019 (a copy of which may be obtained at www.sec.gov and www.sedar.com). Consequently, forward-looking statements should be regarded solely as Zymeworks’ current plans, estimates, and beliefs. Investors should not place undue reliance on forward-looking statements. Zymeworks cannot guarantee future results, events, levels of activity, performance or achievements. Zymeworks does not undertake and specifically declines any obligation to update, republish, or revise any forward-looking statements to reflect new information, future events or circumstances or to reflect the occurrences of unanticipated events, except as may be required by law.
Zymeworks Contacts Investor Inquiries: Ryan Dercho, Ph.D. (604) 678-1388 ir@zymeworks.com
G1 Therapeutics, Inc. (Nasdaq: GTHX), a clinical-stage oncology company, today presented additional findings from a randomized Phase 2 clinical trial demonstrating the myelopreservation benefits of trilaciclib in patients undergoing chemotherapy treatment for 2nd/3rd-line small cell lung cancer (SCLC). Trilaciclib is a first-in-class myelopreservation agent designed to protect the bone marrow from damage by chemotherapy and improve patient outcomes.
The abstract titled “Trilaciclib, a CDK 4/6 inhibitor, mitigates myelosuppression in patients with previously treated extensive-stage small cell lung cancer receiving topotecan” (#8505) was selected for oral presentation at the 2019 American Society of Clinical Oncology (ASCO) Annual Meeting.
In December 2018, the company announced topline data showing that SCLC patients receiving trilaciclib + topotecan (a chemotherapy agent) experienced statistically significant reductions in the duration and occurrence of Grade 4 neutropenia, and that trilaciclib treatment resulted in a reduction in the number of granulocyte colony-stimulating factor (G-CSF) administrations and red blood cell (RBC) transfusions, compared to patients receiving placebo + topotecan. Overall, patients receiving trilaciclib + topotecan showed an improved safety profile compared to those receiving placebo + topotecan.
An analysis of patient-reported outcomes (PRO) data showed clinically meaningful improvements in the treatment experience for patients receiving trilaciclib + topotecan compared to those receiving placebo + topotecan. Patients receiving trilaciclib reported significant improvements in several areas, including: general and physical wellbeing, quality-of-life (QoL) measures specific to lung cancer patients, symptoms and impact of fatigue, and symptoms and effects on physical and functional wellbeing due to anemia.
“Chemotherapy is an effective treatment option for those with cancer. As a treating physician, I regularly see how it also negatively impacts patient health and quality of life. Chemotherapy often causes bone marrow damage that can result in anemia and neutropenia, subsequently leaving patients with severe fatigue and at increased risk of infection,” said Lowell Hart, M.D., Scientific Director of Research, Florida Cancer Specialists and trilaciclib clinical trial investigator. “It was encouraging to observe that in this trial, use of trilaciclib made chemotherapy safer, reducing the rates of chemotherapy-related side effects and the use of rescue interventions commonly used to treat them. Importantly, patient-reported outcomes data showed that the myelopreservation benefits of trilaciclib improved their overall experience on chemotherapy.”
Key findings of the trial included:
Patients receiving trilaciclib + topotecan demonstrated statistically significant reductions in both of the trial’s primary endpoints compared to patients receiving placebo + topotecan: duration of Grade 4 neutropenia in cycle 1 and occurrence of Grade 4 neutropenia.
Trilaciclib treatment reduced the number of G-CSF administrations per cycle and the number of RBC transfusions (on/after week 5) per week compared to placebo.
PRO data showed clinically meaningful improvements in the treatment experience of patients receiving trilaciclib + topotecan compared to those who received placebo + topotecan.
Anti-tumor efficacy measures of overall response rate (ORR), progression-free survival (PFS) and overall survival (OS) were comparable between the trilaciclib + topotecan and placebo + topotecan arms.
Consistent with the three other randomized Phase 2 trials, trilaciclib was well tolerated and there were fewer ≥ Grade 4 treatment-emergent adverse events (TEAEs) in the trilaciclib arm compared to the placebo arm.
Following meetings with U.S and European regulatory authorities to review data from three randomized, placebo-controlled SCLC clinical trials, including data presented at the 2019 ASCO Annual Meeting, the company announced plans to submit marketing applications for trilaciclib for myelopreservation in SCLC. The company expects to file a New Drug Application (NDA) with the U.S. Food and Drug Administration (FDA) and a Marketing Authorization Application (MAA) with the European Medicines Agency (EMA) in 2020.
About Chemotherapy and Trilaciclib Chemotherapy is an effective and important treatment against cancer. However, chemotherapy does not differentiate between healthy cells and cancer cells, killing both, including important stem cells in the bone marrow that produce white blood cells (WBCs), RBCs and platelets. This chemotherapy-induced bone marrow damage is known as myelosuppression. When WBCs, RBCs and platelets become depleted, chemotherapy patients are at increased risk of infection, experience anemia and fatigue, and are at increased risk of bleeding. Myelosuppression often requires the administration of rescue interventions such as growth factors and blood or platelet transfusions, and may also result in chemotherapy dose delays and reductions.
Trilaciclib is a first-in-class myelopreservation agent designed to protect the bone marrow from damage by chemotherapy and improve patient outcomes. G1 expects to submit marketing applications in the U.S. and Europe for trilaciclib for myelopreservation in small cell lung cancer in 2020, and plans to initiate new label expansion trials in 2020.
About G1 Therapeutics G1 Therapeutics, Inc. is a clinical-stage biopharmaceutical company focused on the discovery, development and delivery of innovative therapies that improve the lives of those affected by cancer. The company is advancing three clinical-stage programs. Trilaciclib and lerociclib are designed to enable more effective combination treatment strategies and improve patient outcomes across multiple oncology indications. G1T48 is a potential best-in-class oral selective estrogen receptor degrader (SERD) for the treatment of ER+ breast cancer. G1 also has an active discovery program focused on cyclin-dependent kinase targets.
Forward-Looking Statements This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “may,” “will,” “expect,” “plan,” “anticipate,” “estimate,” “intend” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. Forward-looking statements in this news release include, but are not limited to, the therapeutic potential of trilaciclib, lerociclib and G1T48 and the timing for next steps with regard to the trilaciclib marketing applications, and are based on the Company’s expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Factors that may cause the Company’s actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in the Company’s filings with the U.S. Securities and Exchange Commission, including the “Risk Factors” sections contained therein and include, but are not limited to, the Company’s ability to complete clinical trials for, obtain approvals for and commercialize any of its product candidates; the Company’s initial success in ongoing clinical trials may not be indicative of results obtained when these trials are completed or in later stage trials; the inherent uncertainties associated with developing new products or technologies and operating as a development-stage company; the Company’s development of a CDK4/6 inhibitor to reduce chemotherapy-induced myelosuppression is novel, unproven and rapidly evolving and may never lead to a marketable product; and market conditions. Except as required by law, the Company assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
VANCOUVER, British Columbia–(BUSINESS WIRE)– Zymeworks Inc. (NYSE/TSX: ZYME), a clinical-stage biopharmaceutical company developing multifunctional biotherapeutics, today announced that it has earned a milestone payment from Merck (known as MSD outside the US and Canada) achieved under the companies’ 2014 research and license agreement. Zymeworks will receive US$2.0 million in connection with Merck’s completion of a late-stage preclinical study for a bispecific antibody candidate using Zymeworks’ proprietary Azymetric™ and EFECT™ therapeutic platforms.
“Merck’s completion of this milestone is great news,” said Ali Tehrani, Ph.D., President and CEO of Zymeworks. “We believe this accomplishment from another one of our long-term partners provides additional evidence of the potential of our bispecific antibody technology. We expect similar advancements from our other partners’ therapeutic programs in the years ahead.”
Under the terms of the research and license agreement, Zymeworks has granted Merck a worldwide, royalty-bearing license to research, develop and commercialize certain bispecific therapeutic candidates toward Merck’s therapeutic targets for which Zymeworks is eligible to receive additional development and commercial milestone payments as well as tiered royalties on product sales.
About the Azymetric™ Platform
The Azymetric platform enables the transformation of monospecific antibodies into bispecific antibodies, giving the antibodies the ability to simultaneously bind two different targets. Azymetric bispecific technology enables the development of multifunctional biotherapeutics that can block multiple signaling pathways, recruit immune cells to tumors, enhance receptor clustering degradation, and increase tumor-specific targeting. These features are intended to enhance efficacy while reducing toxicities and the potential for drug resistance. Azymetric bispecifics have been engineered to retain the desirable drug-like qualities of naturally occurring antibodies, including low immunogenicity, long half-life and high stability. In addition, they are compatible with standard manufacturing processes with high yields and purity, potentially significantly reducing drug development costs and timelines.
About the EFECT™ Platform
The EFECT platform is a library of antibody Fc modifications engineered to modulate the activity of the antibody-mediated immune response, which includes both the up- and down-regulation of effector functions. This platform, which is compatible with traditional monoclonal as well as Azymetric bispecific antibodies, further enables the customization of therapeutic responses for different diseases.
About Zymeworks Inc.
Zymeworks is a clinical-stage biopharmaceutical company dedicated to the development of next-generation multifunctional biotherapeutics. The Company’s suite of therapeutic platforms and its fully integrated drug development engine enable precise engineering of highly differentiated product candidates. Zymeworks’ lead clinical candidate, ZW25, is a novel Azymetric™ bispecific antibody currently in Phase 2 clinical development. The Company’s second clinical candidate, ZW49, is a bispecific antibody-drug conjugate currently in Phase 1 clinical development and combines the unique design and antibody framework of ZW25 with Zymeworks’ proprietary ZymeLink™ cytotoxic payload. Zymeworks is also advancing a deep preclinical pipeline in immuno-oncology and other therapeutic areas. In addition, its therapeutic platforms are being leveraged through strategic partnerships with nine biopharmaceutical companies.
This press release includes “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. Forward-looking statements in this news release include, but are not limited to, statements that relate to future development activities in accordance with the terms of Zymeworks’ agreements with Merck and other corporate partners, advancements in Zymeworks’ partners therapeutic programs, potential payments and/or royalties payable to Zymeworks under these agreements, the speed and outcome of drug development plans, Zymeworks’ potential global growth, and other information that is not historical information. When used herein, words such as “enable”, “plan”, “expect”, “will”, “may”, “eligible to”, and similar expressions are intended to identify forward-looking statements. In addition, any statements or information that refer to expectations, beliefs, plans, projections, objectives, performance or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking. All forward-looking statements are based upon Zymeworks’ current expectations and various assumptions. Zymeworks believes there is a reasonable basis for its expectations and beliefs, but they are inherently uncertain. Zymeworks may not realize its expectations, and its beliefs may not prove correct. Actual results could differ materially from those described or implied by such forward-looking statements as a result of various factors, including, without limitation, market conditions and the factors described under “Risk Factors” in Zymeworks’ Quarterly Report on Form 10-Q for its fiscal quarter ended March 31, 2019 (a copy of which may be obtained at www.sec.gov and www.sedar.com). Consequently, forward-looking statements should be regarded solely as Zymeworks’ current plans, estimates and beliefs. Investors should not place undue reliance on forward-looking statements. Zymeworks cannot guarantee future results, events, levels of activity, performance or achievements. Zymeworks does not undertake and specifically declines any obligation to update, republish, or revise any forward-looking statements to reflect new information, future events or circumstances or to reflect the occurrences of unanticipated events, except as may be required by law.
VANCOUVER, British Columbia — Zymeworks Inc. (NYSE/TSX: ZYME), a clinical-stage biopharmaceutical company developing multifunctional biotherapeutics, today announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to ZW25, a novel Azymetric™bispecific antibody, for the first-line treatment of patients with HER2-overexpressing gastroesophageal adenocarcinoma in combination with standard of care chemotherapy.
“Receipt of Fast Track designation from the FDA emphasizes the large unmet need of patients with these types of HER2-expressing cancers,” said Ali Tehrani, Ph.D., Zymeworks’ President & CEO. “This designation is key to our objective of getting important new therapies to patients as quickly as possible. We are pleased with the discussions we’ve had with the FDA thus far and will continue to work together on other opportunities to accelerate the development of ZW25 in additional indications.”
The FDA’s Fast Track designation is designed to facilitate development and expedite the review of therapies to treat a serious diseases where there is a significant unmet medical need. A therapeutic that receives Fast Track designation is eligible for frequent communication with the FDA, accelerated approval and priority review, and rolling submission, which means that a company can submit completed sections of its New Drug Application (NDA) for review by FDA, rather than waiting until every section of the NDA is completed before the entire application can be submitted for review. The primary objective of this program is to accelerate patient access to new and potential lifesaving therapies.
About the Trial
The Phase 2 trial is a two-part open-label study. The primary objectives of this trial are to confirm the safety, tolerability, and anti-tumor activity of ZW25 in combination with global standard of care regimens for gastroesophageal adenocarcinoma, including platinum and fluoropyrimidine-based regimens.
About ZW25
ZW25 is being evaluated in Phase 1 and Phase 2 clinical trials across North America and South Korea. It is a bispecific antibody, based on Zymeworks’ Azymetric™ platform, that can simultaneously bind two non-overlapping epitopes of HER2, known as biparatopic binding. This unique design results in multiple mechanisms of action including dual HER2 signal blockade, increased binding and removal of HER2 protein from the cell surface, and potent effector function leading to encouraging anti-tumor activity in patients. Zymeworks is developing ZW25 as a HER2-targeted treatment option for patients with any solid tumor that expresses HER2. The FDA has granted Fast Track designation to ZW25 for first-line gastroesophageal adenocarcinoma in combination with standard of care chemotherapy and Orphan Drug designation to ZW25 for the treatment of both gastric and ovarian cancers.
About the Azymetric™ Platform
The Azymetric platform enables the transformation of monospecific antibodies into bispecific antibodies, giving the antibodies the ability to simultaneously bind two different targets. Azymetric bispecific technology enables the development of multifunctional biotherapeutics that can block multiple signaling pathways, recruit immune cells to tumors, enhance receptor clustering degradation, and increase tumor-specific targeting. These features are intended to enhance efficacy while reducing toxicities and the potential for drug-resistance. Azymetric bispecifics have been engineered to retain the desirable drug-like qualities of naturally occurring antibodies, including low immunogenicity, long half-life, and high stability. In addition, they are compatible with standard manufacturing processes with high yields and purity, potentially significantly reducing drug development costs and timelines.
About Zymeworks Inc.
Zymeworks is a clinical-stage biopharmaceutical company dedicated to the development of next-generation multifunctional biotherapeutics. Zymeworks’ suite of therapeutic platforms and its fully integrated drug development engine enable precise engineering of highly differentiated product candidates. Zymeworks’ lead clinical candidate, ZW25, is a novel Azymetric™ bispecific antibody currently in Phase 2 clinical development. Zymeworks’ second clinical candidate, ZW49, is a bispecific antibody-drug conjugate currently in Phase 1 clinical development and combines the unique design and antibody framework of ZW25 with Zymeworks’ proprietary ZymeLink™ cytotoxic payload. Zymeworks is also advancing a deep preclinical pipeline in immuno-oncology and other therapeutic areas. In addition, its therapeutic platforms are being leveraged through strategic partnerships with nine biopharmaceutical companies.
This press release includes “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. Forward-looking statements in this news release include, but are not limited to, the speed and outcome of drug development plans, the speed, frequency and outcome of communications with the FDA, patient access to ZW25, and other information that is not historical information. When used herein, words such as “enable”, “plan”, “expect”, “will”, “may”, “eligible”, “continue to” and similar expressions are intended to identify forward-looking statements. In addition, any statements or information that refer to expectations, beliefs, plans, projections, objectives, performance or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking. All forward-looking statements are based upon Zymeworks’ current expectations and various assumptions. Zymeworks believes there is a reasonable basis for its expectations and beliefs, but they are inherently uncertain. Zymeworks may not realize its expectations, and its beliefs may not prove correct. Actual results could differ materially from those described or implied by such forward-looking statements as a result of various factors, including, without limitation, market conditions and the factors described under “Risk Factors” in Zymeworks’ Quarterly Report on Form 10-Q for its fiscal quarter ended March 31, 2019 (a copy of which may be obtained at www.sec.gov and www.sedar.com). Consequently, forward-looking statements should be regarded solely as Zymeworks’ current plans, estimates and beliefs. Investors should not place undue reliance on forward-looking statements. Zymeworks cannot guarantee future results, events, levels of activity, performance or achievements. Zymeworks does not undertake and specifically declines any obligation to update, republish, or revise any forward-looking statements to reflect new information, future events or circumstances or to reflect the occurrences of unanticipated events, except as may be required by law.
Zymeworks Inc. Contacts Investor Inquiries: Ryan Dercho, Ph.D. (604) 678-1388 ir@zymeworks.com
Commercial leader brings recent migraine space experience to Satsuma as it advances lead product candidate STS101 into Phase 3 development
SOUTH SAN FRANCISCO, Calif., May 29, 2019 /PRNewswire/ — Satsuma Pharmaceuticals, Inc. (“Satsuma” or “the Company”), a Phase 3-stage biopharmaceutical company developing STS101, (dihydroergotamine (DHE) nasal powder) for the acute treatment of migraine, today announced that Elisabeth Sandoval has been appointed as a member of Satsuma’s Board of Directors, effective immediately. Ms. Sandoval most recently served as Chief Commercial Officer and Executive Vice President of Corporate Strategy for Alder Biopharmaceuticals, an NDA-stage company focused on developing novel therapeutic antibodies for the treatment of migraine, and previously served as Chief Commercial Officer of KYTHERA Biopharmaceuticals.
“We welcome Elisabeth to the Satsuma Board. Her extensive drug product commercialization experience, including recent work in the migraine field, and her leadership and strategy skills, will greatly benefit Satsuma as the Company advances STS101 through Phase 3 development and toward commercialization,” commented Heath Lukatch, Ph.D., Chairman of the Board of Directors of Satsuma Pharmaceuticals.
As Chief Commercial Officer and Executive Vice President of Corporate Strategy at Alder Biopharmaceuticals, Ms. Sandoval was responsible for developing and leading the company’s commercial, strategy, medical affairs, investor relations, corporate communications, and business development functions. Before joining Alder, she was Chief Commercial Officer for KYTHERA Biopharmaceuticals, where she led the commercial strategy and execution including the hiring and development of all commercial functions and launch of a first-in-class product prior to KYTHERA’s acquisition by Allergan. Before KYTHERA, Ms. Sandoval was Vice President of Marketing for Bausch and Lomb Surgical, leading all marketing strategy and execution globally. Prior to this position, Ms. Sandoval was Vice President of Global Marketing at Allergan with responsibility for developing the global commercial strategy for the Medical Aesthetics division and execution of key product launches. She spent 23 years at Allergan in sales and marketing leadership roles in the specialties of dermatology, neurology, and aesthetics. Ms. Sandoval serves on the board of directors for Menlo Therapeutics and ALASTIN Skincare, a privately held company. Ms. Sandoval began her career in research and development at Johnson & Johnson’s Ethicon division. She holds an MBA from Pepperdine University and a B.S. in Biology from the University of California, Irvine.
About Satsuma Pharmaceuticals Satsuma Pharmaceuticals is a Phase 3-stage biopharmaceutical company focused on developing STS101 as an important and differentiated therapeutic option for the acute treatment of migraine. STS101 is a novel and proprietary investigational drug-device combination product specifically designed to enable intranasal administration of the anti-migraine drug, dihydroergotamine (DHE), with a pharmacokinetic profile optimized to provide consistent and robust clinical efficacy. In developing STS101, Satsuma has applied proprietary nasal drug delivery, dry-powder formulation, and engineered drug particle technologies to create a compact, simple-to-use, self-administered, and non-injectable DHE product. The Company believes STS101 will be an attractive migraine treatment option for many patients and may enable a larger number of people with migraine to realize the long-recognized therapeutic benefits of DHE therapy. STS101 has undergone extensive pre-clinical optimization and recently completed a Phase 1 clinical trial.
Satsuma is headquartered in South San Francisco, California with operations in both California and Research Triangle Park, North Carolina.
Corporate Contacts
Tom O’Neil, Chief Financial Officer Satsuma Pharmaceuticals, Inc. tom@satsumarx.com
John Kollins, President and Chief Executive Officer Satsuma Pharmaceuticals, Inc. john@satsumarx.com
Austin, TX and Montreal, QC (May 28, 2019) – Forbius, a clinical-stage company that develops novel biologics for the treatment of cancer and fibrosis, announced today that it has obtained approval from Health Canada to conduct Phase 2 clinical trials evaluating AVID100, a novel anti-EGFR antibody-drug conjugate (ADC), in EGFR-overexpressing squamous cell carcinoma of the head and neck (SCCHN), squamous non-small cell lung cancer (sqNSCLC) and triple-negative breast cancer (TNBC).
Expansion of ongoing AVID100 Phase 2 trials in patients with SCCHN, sqNSCLC and TNBC who overexpress EGFR to include clinical sites in Canada
AVID100 is the most advanced and broadly active anti-EGFR ADC in clinical development, targeting both wild-type and mutant forms of EGFR
Recommended Phase 2 dose (RP2D) of 220 mg/m2 (~6 mg/kg) q3w, one of the highest amongst ADCs in development and predicted to be in therapeutic range
AVID100-01 (NCT03094169) is a Phase 2, open label, multicenter study to evaluate the safety and efficacy of AVID100 in up to 100 SCCHN, sqNSCLC and TNBC patients with confirmed EGFR-overexpression. The trial is currently enrolling patients at centers in the U.S. and will be expanding to additional clinical sites in Canada.
Preclinical data demonstrated AVID100 to be highly potent and selectively cytotoxic against EGFR-expressing cancer cells while sparing normal keratinocytes. A Phase 1 dose-escalation study in patients with advanced solid tumors of epithelial origin confirmed that AVID100 was well tolerated and established an RP2D of 220 mg/m2 (~6 mg/kg), which is expected to be in the therapeutically active range and is one of the highest RP2Ds reported for ADCs with maytansinoid payload.
Approximately 20–25% of patients with SCCHN, sqNSCLC and TNBC have tumors that highly overexpress EGFR. No targeted therapy is approved for these indications with confirmed EGFR-overexpression.
About AVID100 and the AVID100-01 Trial
AVID100 is a highly potent EGFR-targeting antibody-drug conjugate (ADC) engineered to achieve enhanced anti-tumor efficacy without a corresponding increase in toxicity against skin and other EGFR-expressing normal tissues. In preclinical studies, AVID100 demonstrated significant anti-cancer activity in EGFR-overexpressing tumor models resistant to marketed EGFR inhibitors. AVID100 is the most advanced, broadly active anti-EGFR ADC in clinical development and targets both wild-type and mutant forms of EGFR.
AVID100-01 (NCT03094169) is an open-label, multicenter, dose-expansion study to evaluate the efficacy, safety and tolerability of AVID100 in patients with confirmed EGFR-overexpressing sqNSCLC (IHC 3+), SCCHN (IHC 3+) and TNBC (IHC 2+/3+) (more than 50% of cells with EGFR 3+ or more than 75% of cells with EGFR 2+ staining).
About Forbius: Targeting TGF-beta and EGFR Pathways in Fibrosis and Cancer
Forbius is a clinical-stage protein engineering company that designs and develops novel biologics for the treatment of fibrosis and cancer. Our current focus is the development of agents targeting the transforming growth factor-beta (TGF-beta) and epidermal growth factor receptor (EGFR) pathways.
MONTRÉAL and BOSTON, May 29, 2019 — enGene Inc., the high growth biotechnology company developing the Gene Pill™ – a robust, proprietary non-viral vector platform to deliver gene therapies via oral administration – today announced the appointment of José M. Lora, Ph.D., as Chief Scientific Officer. Dr. Lora will join the executive management team and will be responsible for setting the scientific strategy and priorities for enGene, as well as overseeing day-to-day research operations.
“We are thrilled to add José’s deep scientific expertise and talent to enGene’s leadership team,” said Jason Hanson, Chief Executive Officer and President of enGene. “Along with his scientific prowess comes his deep drive to bring forward meaningful medications to impact patients’ lives. He is truly the ideal addition to our team.”
“The potential for enGene’s non-viral vector platform to create transformational medicines is remarkably exciting to me personally,” stated Dr. Lora. “I am honored to help build upon the great successes already achieved by enGene’s talented team and to our advancement of promising programs for the benefit of patients.”
Dr. Lora brings to enGene an extensive track record of leading discovery programs from concept to clinic across various therapeutic areas and modalities. He has been intimately involved in numerous IND-filings, is a co-inventor on 10+ patents and has co-authored 40+ peer-reviewed publications and book chapters. Dr. Lora joins enGene from Synlogic, Inc., where he served as Vice President of Research. In this role, José was responsible for building the Immunomodulation Therapy Area and for immuno-oncology programs from discovery up to human proof-of-mechanism/proof-of-concept. Prior to joining Synlogic, José ascended in positions of increasing responsibility at Constellation Pharmaceuticals, Inc., where he was ultimately responsible for establishing and managing immunomodulatory and immuno-epigenetics functions. Early in his career, José led efforts in immunology, liver fibrosis and respiratory diseases (including COPD and Cystic Fibrosis) at Millennium Pharmaceuticals, Inc., Roche Palo Alto, LLC, and GlaxoSmithKline. Dr. Lora obtained his Ph.D. in Cellular and Molecular Biology at CSIC/University of Sevilla, Spain, and completed his post-doctoral studies at Brown University and the University of Utah in the USA.
“I look forward to working with José as enGene enters its exciting new phase of growth,” said Dr. Cheung, Co-founder, and Chief Technology Officer at enGene. “We are proud that the promise of our platform and the milestones our team has accomplished have attracted an industry veteran of José’s caliber to join our journey.”
About enGene Inc. enGene Inc. is a biotechnology company developing a proprietary non-viral gene therapy platform for localized delivery of nucleic acid payloads to mucosal tissues. The dually derived chitosan (DDX®) platform has a high-degree of payload flexibility including DNA and various forms of RNA (siRNA, shRNA, lncRNA, etc.) with broad tissue and disease applications. In addition to developing gene therapies for mucosal tissues such as the bladder, enGene has developed a unique gut-optimized gene delivery formulation into an orally available Gene Pill™ to provide oral delivery of a wide range of protein drugs. Oral gene delivery is a revolutionary improvement over current protein-drug delivery methods which involve injections and represent risks of side effects, high cost, poor patient compliance and systemic problems due to the need to administer higher doses. The company is evolving its technology to enable treatments of all mucosal tissues such as the lung and eye among others.
For further information:
Jason Hanson, CEO and President, enGene Inc., jhanson@engene.com
VANCOUVER, British Columbia–(BUSINESS WIRE)–Zymeworks Inc. (NYSE/TSX: ZYME), a clinical-stage biopharmaceutical company developing multifunctional biotherapeutics, today announced that it has entered into a licensing agreement that grants Iconic Therapeutics, Inc. (Iconic) non-exclusive rights to Zymeworks’ proprietary ZymeLink™ antibody-drug conjugate (ADC) platform for the development of its ICON-2 Tissue Factor ADC for cancer. This is the first collaboration leveraging the ZymeLink platform and represents Zymeworks’ third technology platform licensed to a collaborator.
“We believe this first ZymeLink licensing deal provides further validation of our novel ADC technology, which is already being used in Zymeworks’ own clinical candidate, ZW49.”
– Ali Tehrani, Ph.D., President and CEO, Zymeworks
“We believe this first ZymeLink licensing deal provides further validation of our novel ADC technology, which is already being used in Zymeworks’ own clinical candidate, ZW49,” said Ali Tehrani, Ph.D., President and Chief Executive Officer of Zymeworks. “Historically, traditional ADC development has been plagued by a number of challenges related to toxicity and efficacy. Our research has shown that ZymeLink has the capacity to significantly enhance exposure and tolerability, broadening the therapeutic window and leading to potentially safer and more efficacious therapeutic candidates.”
“Zymeworks’ technology provides properties and capabilities we believe will enhance and leverage Iconic’s Tissue Factor platform,” commented William Greene, M.D., Iconic’s Chief Executive Officer. “Having evaluated several alternatives, we are confident that we can develop a truly differentiated ADC with ZymeLink. Tissue Factor is an important target in solid tumors, and we believe the combination of our best-in-class antibodies with Zymeworks’ next generation payload technology will deliver an ADC with enhanced safety and efficacy with the potential to be an important addition to the cancer armamentarium. We look forward to progressing ICON-2 to the clinic in 2020 and more broadly, to further developing our pipeline of therapeutic approaches to targeting Tissue Factor mediated diseases.”
Under the terms of the agreement, Zymeworks will be eligible to receive development and commercial milestone payments and tiered royalties on worldwide net sales. The agreement also provides Zymeworks co-promotion rights with increased royalties for products developed using the Iconic ADC program. If Iconic outlicenses the program, in lieu of co-promotion rights, Zymeworks will receive a share of the revenue Iconic receives from any partners as well as tiered royalties on worldwide net sales.
About the ZymeLink™ Platform
The ZymeLink platform is a set of proprietary cytotoxic drugs and linkers designed to create stable, polar ADCs for the targeted delivery of therapeutics with significantly enhanced exposure and tolerability leading to increased efficacy against targets that traditionally have been challenging for ADCs. The ZymeLink platform is compatible with monoclonal and bispecific antibodies and is intended to facilitate the development of next-generation antibody-drug conjugates with broad therapeutic windows.
About Iconic Therapeutics
Iconic Therapeutics is a venture-backed biopharmaceutical company dedicated to translating an understanding of the role of Tissue Factor biology to new therapeutics for diseases such as macular degeneration and cancer. The company has developed a portfolio of proprietary molecules, which bind to and antagonize Tissue Factor expressed in disease, both in retina and in solid tumors.
About Zymeworks Inc.
Zymeworks is a clinical-stage biopharmaceutical company dedicated to the development of next-generation multifunctional biotherapeutics. The Company’s suite of therapeutic platforms and its fully integrated drug development engine enable precise engineering of highly differentiated product candidates. Zymeworks’ lead clinical candidate, ZW25, is a novel Azymetric™ bispecific antibody currently in Phase 2 clinical development. The Company’s second clinical candidate, ZW49, is a bispecific antibody-drug conjugate currently in Phase 1 clinical development and combines the unique design and antibody framework of ZW25 with Zymeworks’ proprietary ZymeLink™ cytotoxic payload. Zymeworks is also advancing a deep preclinical pipeline in immuno-oncology and other therapeutic areas. In addition, its therapeutic platforms are being leveraged through multiple strategic partnerships with nine biopharmaceutical companies.
This press release includes “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. Forward-looking statements in this news release include, but are not limited to, statements that relate to potential milestone payments, royalties and other revenue, co-promotion rights, ZymeLink’s effect on the ADC field and its potential with respect to therapeutic treatment and development of therapeutic candidates, and other information that is not historical information. When used herein, words and phrases such as “will,” “eligible to,” “entitled to,” “look forward to,” and similar expressions are intended to identify forward-looking statements. In addition, any statements or information that refer to expectations, beliefs, plans, projections, objectives, performance or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking. All forward-looking statements are based upon Zymeworks’ current expectations and various assumptions. Zymeworks believes there is a reasonable basis for its expectations and beliefs, but they are inherently uncertain. Zymeworks may not realize its expectations, and its beliefs may not prove correct. Actual results could differ materially from those described or implied by such forward-looking statements as a result of various factors, including, without limitation, market conditions and the factors described under “Risk Factors” in Zymeworks’ Quarterly Report on Form 10-Q for its fiscal quarter ended March 31, 2019 (a copy of which may be obtained at www.sec.gov and www.sedar.com). Consequently, forward-looking statements should be regarded solely as Zymeworks’ current plans, estimates and beliefs. Investors should not place undue reliance on forward-looking statements. Zymeworks cannot guarantee future results, events, levels of activity, performance or achievements. Zymeworks does not undertake and specifically declines any obligation to update, republish, or revise any forward-looking statements to reflect new information, future events or circumstances or to reflect the occurrences of unanticipated events, except as may be required by law.
VICTORIA, British Columbia–(BUSINESS WIRE)– Aurinia Pharmaceuticals Inc. (NASDAQ: AUPH / TSX:AUP) (“Aurinia” or the “Company”) today reported financial results for the first quarter ended March 31, 2019, and provided an update on recent operational highlights. Amounts, unless specified otherwise, are expressed in U.S. dollars.
First Quarter 2019 Highlights
Fully-enrolled AURORA Phase 3 trial in lupus nephritis (“LN”) continues on track with results anticipated in late 2019.
Reported results from a Phase 2a Dry Eye study with voclosporin ophthalmic solution (“VOS”) that achieved statistically superior efficacy in secondary objective endpoints compared to cyclosporin ophthalmic emulsion 0.05% (Restasis®), the current DES market leader. VOS did not meet the primary endpoint as both drugs were well tolerated and demonstrated less than anticipated drop discomfort.
Received a Notice of Allowance from the United States Patent and Trademark Office (“USPTO”) for claims which have the potential to cover voclosporin’s method of use and dosing protocol for lupus nephritis (“LN’) until December 2037.
Appointed Mr. Peter Greenleaf as Chief Executive Officer and Dr. George Milne to Chairman of the Board of Directors.
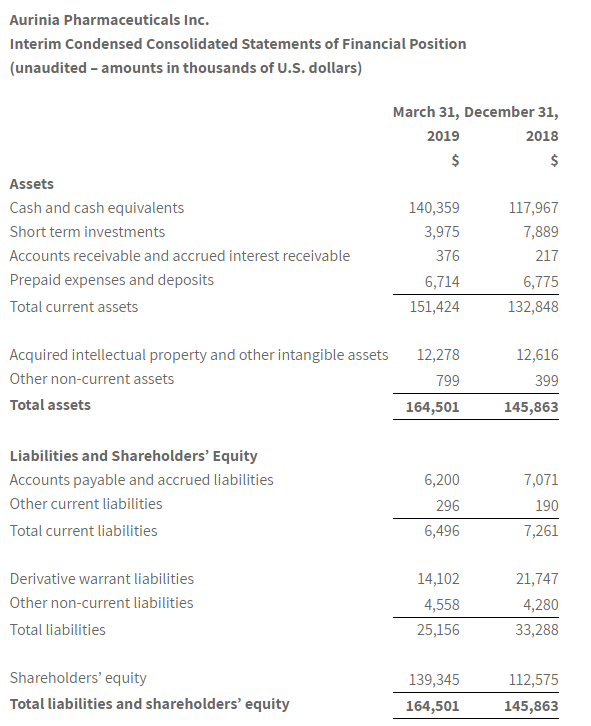
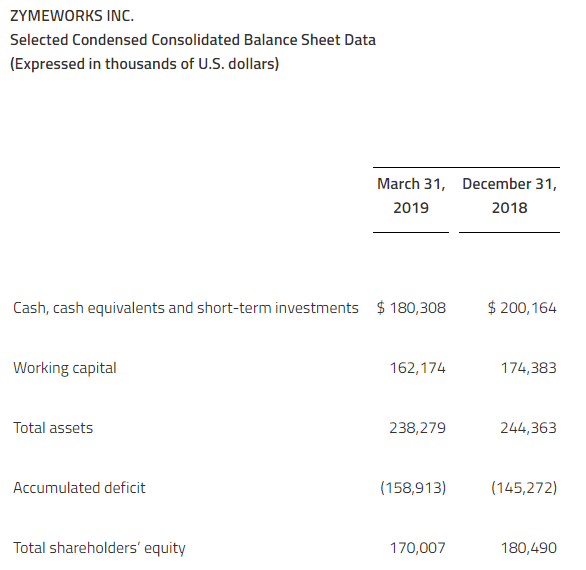
Cash, cash equivalents, and short-term investments of $144.3 million as of March 31, 2019.
Recent Director and Officer Appointments
On April 29, 2019, Aurinia appointed Peter Greenleaf as Chief Executive Officer and a Director on the Aurinia Board.
Concurrently, Dr. Richard M. Glickman, who previously announced his plans to retire on November 6, 2018, stepped down from his role as Chairman and CEO. Dr. Glickman remains an advisor to the Company for a period of 12 months.
“It is an honor to join Aurinia at this time and lead the organization through its next phase of growth to advance voclosporin toward commercialization based upon the Phase 3 AURORA results in lupus nephritis anticipated by the end of this year,” commented Mr. Peter Greenleaf, Chief Executive Officer of Aurinia. “After following the Aurinia story and after conducting further due diligence, I am truly impressed with the Aurinia team, their ability to execute and advance voclosporin in a cost-efficient manner, with the ongoing goal of bringing voclosporin to help patients suffering from LN.”
In conjunction with Dr. Glickman’s retirement as the Chairman, the Board elevated George M. Milne, Jr., Ph.D., to the position of Chairman of the Board effective April 29, 2019. In addition, the Board appointed Dr. Daniel Billen to the Board also effective April 29, 2019.
Dr. Milne stated, “With the appointment of Peter and Daniel to the board, combined with our experienced and committed employees and management team, Aurinia is strongly positioned to achieve our milestones and maximize the value of voclosporin for all of our stakeholders.
VOS for Dry Eye Syndrome (“DES”)
Based upon the exploratory Phase 2a results generated with VOS in a head-to-head comparison vs. the current market leader for the treatment of DES, Aurinia plans to initiate a Phase 2/3 study by late 2019. This study will encompass certain critical regulatory requirements that the FDA has traditionally required for DES product approval, these requirements include both dose-optimization requirements along with a comparison versus vehicle.
“I’m confident that the internal Aurinia team along with our key ophthalmology clinical advisors have crafted a framework of a plan that minimizes the clinical and regulatory risk for VOS and maximizes our probability of launching VOS into the multi-billion dollar DES market in due course,” said Michael R. Martin, Chief Operating Officer of Aurinia.
Financial Liquidity at March 31, 2019
As at March 31, 2019, Aurinia had cash, cash equivalents and short-term investments of $144.3 million compared to $125.9 million of cash, cash equivalents and short-term investments as at December 31, 2018. Net cash used in operating activities was $13.1 million for the first quarter ended March 31, 2019, compared to $14.4 million for the first quarter ended March 31, 2018.
The Company believes, that based on its current plans that Aurinia has sufficient financial resources to fund the existing LN program, including the AURORA trial and the AURORA 2 extension trial, complete the NDA submission to the FDA, conduct the ongoing Phase 2 study for FSGS, commence additional DES studies and fund operations into mid-2020.
The increase in our cash position at March 31, 2019, was primarily the result of the following:
At-The-Market (“ATM”) Facility
On November 30, 2018, Aurinia had entered into an open market sale agreement with Jefferies LLC pursuant to which the Company could from time to time sell, through ATM offerings, common shares that would have an aggregate offering amount of up to $30 million. The ATM was fully utilized in the first quarter. Aurinia received gross proceeds of $30 million and issued 4.6 million common shares. The Company incurred share issue costs of $1.2 million including a 3% commission and professional and filing fees related to the ATM offerings.
February 14, 2014 Warrant Exercises
The remaining derivative warrants outstanding from the February 14, 2014, private placement were exercised in the first quarter ended March 31, 2019. Certain holders of these warrants elected the cashless exercise option and the Company issued 687,000 common shares on the cashless exercise of 1.3 million warrants. Three holders of 464,000 warrants exercised these warrants for cash, at a price of $3.2204 per common share. The Company received cash proceeds of $1.5 million and issued 464,000 common shares.
Financial Results for the First Quarter Ended March 31, 2019
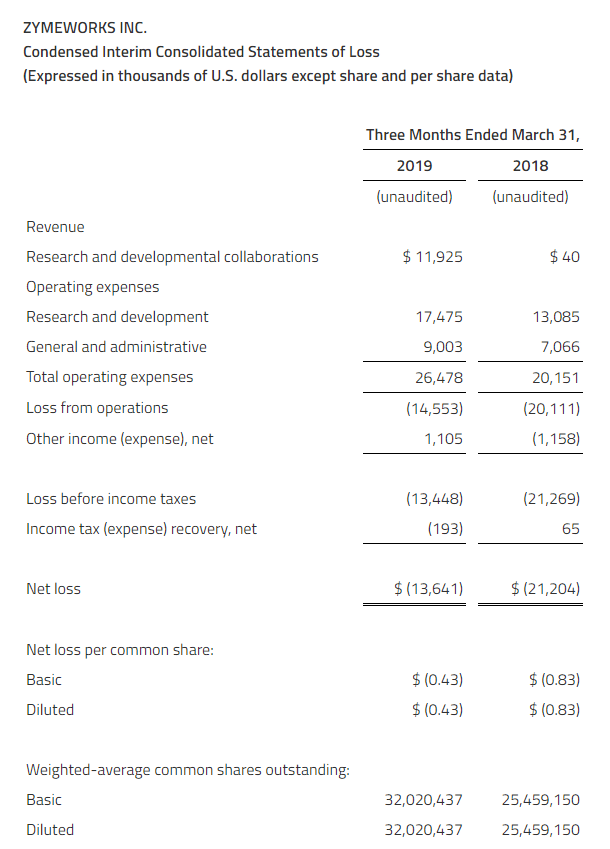
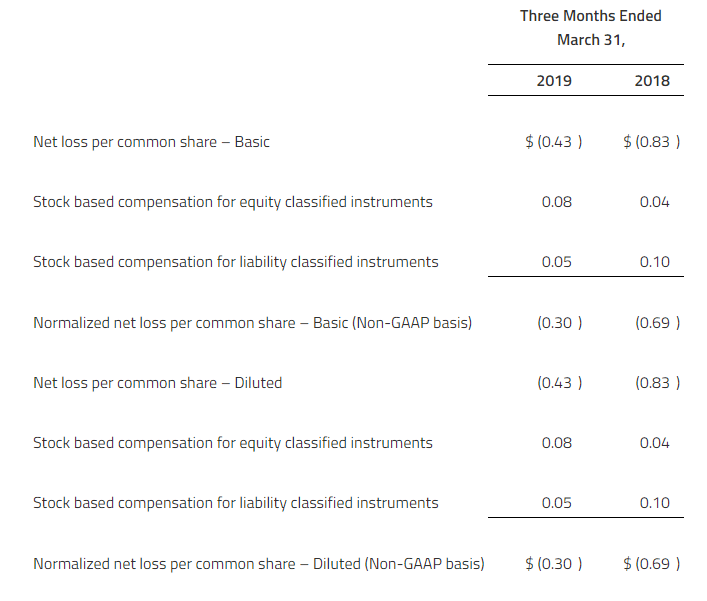
The Company reported a consolidated net loss of $12.4 million or $0.14 per common share for the first quarter ended March 31, 2019, as compared to a consolidated net loss of $15.5 million or $0.18 per common share for the first quarter ended March 31, 2018.
The loss for the first quarter ended March 31, 2019, reflected a reduction of $1.7 million in the estimated fair value of derivative warrant liabilities compared to an increase of $2.6 million in the estimated fair value of derivative warrant liabilities for the first quarter ended March 31, 2018. The derivative warrant liabilities will ultimately be eliminated on the exercise or forfeiture of the warrants and will not result in any cash outlay by the Company.
The loss before the change in estimated fair value of derivative warrant liabilities and income tax was $14.1 million for the first quarter ended March 31, 2019, compared to $12.9 million for the same period in 2018.
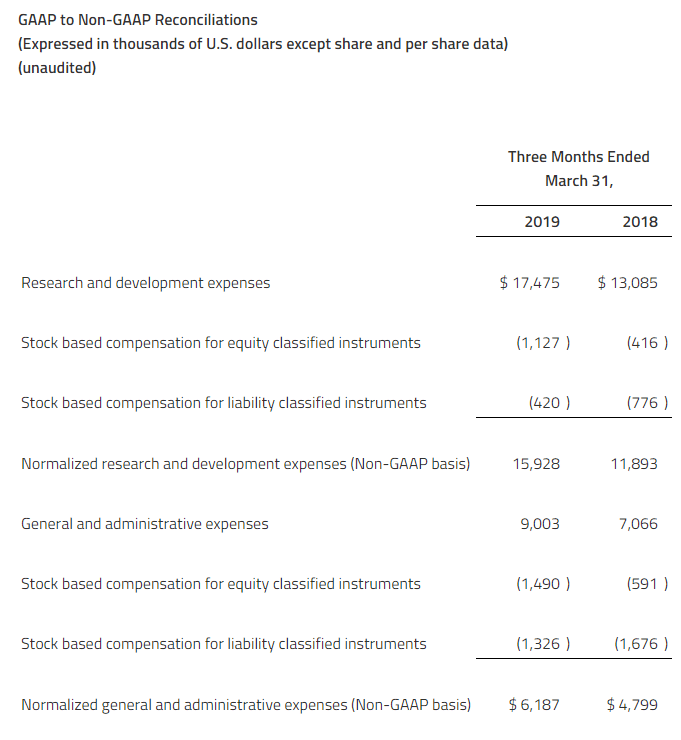
Research and development (“R&D”) expenses increased to $10.6 million for the first quarter ended March 31, 2019, compared to $8.9 million for the first quarter ended March 31, 2018. The increase in these expenses primarily reflected completion costs for the DES study and higher costs incurred for the AURORA 2 extension trial, the DDI study and the FSGS Phase 2a study as these studies had more activity in the first quarter ended March 31, 2019, compared to the same period in 2018.
Corporate, administration and business development expenses increased slightly to $3.9 million for the first quarter of 2019, compared to $3.8 million for the first quarter of 2018.
This press release should be read in conjunction with our unaudited interim condensed consolidated financial statements and the Management’s Discussion and Analysis for the first quarter ended March 31, 2019, which are accessible on Aurinia’s website, on SEDAR or on EDGAR.
Aurinia will host a conference call and webcast to discuss the first quarter ended March 31, 2019, financial results today, Monday, May 13, 2019, at 4:30 p.m. ET. This event can be accessed on the investor section of the Aurinia website.
About Aurinia
Aurinia Pharmaceuticals is a late clinical-stage biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are impacted by serious diseases with a high unmet medical need. The Company is currently developing an investigational drug, for the treatment of Lupus Nephritis, Focal Segmental Glomerulosclerosis, and Dry Eye Syndrome. The Company’s head office is in Victoria, British Columbia and focuses its development efforts globally.
About Voclosporin
Voclosporin, an investigational drug, is a novel and potentially best-in-class calcineurin inhibitor (“CNI”) with clinical data in over 2,600 patients across indications. Voclosporin is an immunosuppressant, with a synergistic and dual mechanism of action. By inhibiting calcineurin, voclosporin blocks IL-2 expression and T-cell mediated immune responses and stabilizes the podocyte in the kidney. It has been shown to have a more predictable pharmacokinetic and pharmacodynamic relationship (potentially requires no therapeutic drug monitoring), an increase in potency (vs cyclosporin), and an improved metabolic profile compared to legacy CNIs. Aurinia anticipates that upon regulatory approval, patent protection for voclosporin will be extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act and comparable laws in other countries and until April 2028 with anticipated pediatric extension. Further, the new Notice of Allowanceis expected to result in the issuance of a U.S. patent with a term extending to December 2037. If the FDA approves the use of voclosporin for LN and the label for such use follows the dosing protocol under the Notice of Allowance, the issuance of this patent will expand the scope of intellectual property protection for voclosporin to December 2037.
About VOS
Voclosporin ophthalmic solution (“VOS”) is an aqueous, preservative-free nanomicellar solution intended for use in the treatment of DES. A Phase 2a study was recently completed with results released in January of 2019. Previously, a Phase 1 study with healthy volunteers and patients with DES was also completed as were studies in rabbit and dog models. VOS has IP protection until 2031.
About LN
Lupus Nephritis (“LN”) in an inflammation of the kidney caused by Systemic Lupus Erythematosus (“SLE”) and represents a serious progression of SLE. SLE is a chronic, complex and often disabling disorder. The disease is highly heterogeneous, affecting a wide range of organs and tissue systems. Unlike SLE, LN has straightforward disease outcomes (measuring proteinuria) where an early response correlates with long-term outcomes. In patients with LN, renal damage results in proteinuria and/or hematuria and a decrease in renal function as evidenced by reduced estimated glomerular filtration rate (“eGFR”), and increased serum creatinine levels. LN is debilitating and costly and if poorly controlled, LN can lead to permanent and irreversible tissue damage within the kidney, resulting in end-stage renal disease (“ESRD”), thus making LN a serious and potentially life-threatening condition.
About FSGS
Focal segmental glomerulosclerosis (“FSGS”) is a rare disease that attacks the kidney’s filtering units (glomeruli) causing serious scarring which leads to permanent kidney damage and even renal failure. FSGS is one of the leading causes of Nephrotic Syndrome (“NS”) and is identified by biopsy and proteinuria. NS is a collection of signs and symptoms that indicate kidney damage, including large amounts of protein in the urine; low levels of albumin and higher than normal fat and cholesterol levels in the blood, and edema. Similar to LN, early clinical response (measured by reduction of proteinuria) is thought to be critical to long-term kidney health in patients with FSGS. Currently, there are no approved therapies for FSGS in the United States and the European Union.
About DES
Dry eye syndrome (“DES”) is characterized by irritation and inflammation that occurs when the eye’s tear film is compromised by reduced tear production, imbalanced tear composition, or excessive tear evaporation. The impact of DES ranges from subtle, yet constant eye irritation to significant inflammation and scarring of the eye’s surface. Discomfort and pain resulting from DES can reduce quality of life and cause difficulty reading, driving, using computers and performing daily activities. DES is a chronic disease. There are currently three FDA approved therapies for the treatment of dry eye; however, there is opportunity for potential improvement in the effectiveness by enhancing tolerability, onset of action and alleviating the need for repetitive dosing.
Forward-Looking Statements
Certain statements made in this press release may constitute forward-looking information within the meaning of applicable Canadian securities law and forward-looking statements within the meaning of applicable United States securities law. These forward-looking statements or information include but are not limited to statements or information with respect to AURORA having data around the end of this year, completing NDA submissions in a successful and timely manner, voclosporin being potentially a best-in-class CNI with robust intellectual property exclusivity; and that Aurinia has sufficient financial resources to fund the existing LN program, including the AURORA trial, and the NDA submission to the FDA, conduct the current Phase 2a study for FSGS, commence additional studies for DES and fund operations into mid-2020 and that the efficacy endpoint clearly signals that VOS has the potential to have a more rapid onset than Restasis® as measured by signs of the disease. It is possible that such results or conclusions may change based on further analyses of these data. Words such as “anticipate”, “will”, “believe”, “estimate”, “expect”, “intend”, “target”, “plan”, “goals”, “objectives”, “may” and other similar words and expressions, identify forward-looking statements. We have made numerous assumptions about the forward-looking statements and information contained herein, including among other things, assumptions about: the market value for the LN & DES programs; that another company will not create a substantial competitive product for Aurinia’s LN and DES business without violating Aurinia’s intellectual property rights; the burn rate of Aurinia’s cash for operations; the costs and expenses associated with Aurinia’s clinical trials; the planned studies achieving positive results; Aurinia being able to extend and protect its patents on terms acceptable to Aurinia; and the size of the LN or DES markets. Even though the management of Aurinia believes that the assumptions made, and the expectations represented by such statements or information are reasonable, there can be no assurance that the forward-looking information will prove to be accurate.
Forward-looking information by their nature are based on assumptions and involve known and unknown risks, uncertainties and other factors which may cause the actual results, performance or achievements of Aurinia to be materially different from any future results, performance or achievements expressed or implied by such forward-looking information. Should one or more of these risks and uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those described in forward-looking statements or information. Such risks, uncertainties and other factors include, among others, the following: difficulties, delays, or failures we may experience in the conduct of our AURORA clinical trial; difficulties we may experience in completing the development and commercialization of voclosporin; the market for the LN business may not be as estimated; Aurinia may have to pay unanticipated expenses; estimated costs for clinical trials may be underestimated, resulting in Aurinia having to make additional expenditures to achieve its current goals; Aurinia not being able to extend or fully protect its patent portfolio for voclosporin; and competitors may arise with similar products. Although we have attempted to identify factors that would cause actual actions, events or results to differ materially from those described in forward-looking statements and information, there may be other factors that cause actual results, performances, achievements or events to not be as anticipated, estimated or intended. Also, many of the factors are beyond our control. There can be no assurance that forward-looking statements or information will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, you should not place undue reliance on forward-looking statements or information.
Except as required by law, Aurinia will not update forward-looking information. All forward-looking information contained in this press release is qualified by this cautionary statement. Additional information related to Aurinia, including a detailed list of the risks and uncertainties affecting Aurinia and its business can be found in Aurinia’s most recent Annual Information Form available by accessing the Canadian Securities Administrators’ System for Electronic Document Analysis and Retrieval (SEDAR) website or the U.S. Securities and Exchange Commission’s Electronic Document Gathering and Retrieval System (EDGAR) website.
MONTREAL, May 14, 2019 — Medexus Pharmaceuticals Inc. (the “Company”) (TSXV: MDP, OTCQB: PDDPF) announced today it has submitted to the TSX Venture Exchange (the “Exchange”) a notice of its intention to make a normal course issuer bid (the “Bid”). Provided the Company receives the approval of the Exchange, the Company may purchase for cancellation, from time to time, as it considers advisable, up to 1,005,333 of its issued and outstanding common shares (“Shares”), being approximately 6.8% of the Company’s currently outstanding Shares and approximately 10% of the Company’s Public Float (as that term is defined in the policies of the Exchange) under the Bid. The Company may not purchase more than 2% of the issued and outstanding Shares during any 30 day period, which as of the date of this announcement represents 294,925 Shares.
Medexus has entered into an “automatic securities purchase plan” (as defined under applicable securities laws) with Canaccord Genuity Corp. (“Canaccord”) for the purpose of making purchases under the Bid (the “ASPP”). Such purchases will be determined by Canaccord in its sole discretion, without consultation with Medexus, having regard to the price limitation and aggregate purchase limitation and other terms of the ASPP and the rules and policies of the Exchange. Conducting the Bid as an ASPP allows Shares to be purchased at times when the Company would otherwise be prohibited from doing so pursuant to securities laws and its internal trading policies.
The maximum number of Shares to be purchased pursuant to the Bid represents approximately 10% of Public Float on the date hereof. Purchases of Shares will be made on the open market through the facilities of the Exchange or by other means as may be permitted by the Exchange (including through other published markets). The price that Medexus will pay for any Shares purchased by it will be the prevailing market price of the Shares on the Exchange at the time of such purchase. The actual number of Shares that may be purchased for cancellation and the timing of any such purchases will be determined by Canaccord in accordance with the ASPP.
It is anticipated that the Bid will commence on or about May 16, 2019 and will conclude on the earlier of (i) May 15, 2020, (ii) the date on which the Company has purchased the maximum number Shares to be acquired pursuant to the Bid, or (iii) the Company providing a notice of termination of the Bid to the TSX-V.
The Board of Directors of Medexus believes that the market price of the Shares may not fully reflect the underlying value of the Shares and that the proposed purchase of Shares would be in the best interests of the Company and is a desirable use of corporate funds. Such purchases will increase the proportionate interest of and may be advantageous to, all remaining shareholders of Medexus. In addition, the purchases by Medexus may increase liquidity to shareholders wishing to sell their Shares. All Shares purchased by the Company will be canceled.
About Medexus
Medexus is a leading specialty pharmaceutical company with a strong North American commercial platform. The Company’s vision is to provide the best healthcare products to healthcare professionals and patients, through our core values of Quality, Innovation, Customer Service and Teamwork. Medexus Pharmaceuticals is focused on the therapeutic areas of auto-immune disease and pediatrics. The leading products are Rasuvo and Metoject, a unique formulation of methotrexate (auto-pen and pre-filled syringe) designed to treat rheumatoid arthritis and other auto-immune diseases; and Rupall, an innovative prescription allergy medication with a unique mode of action.
For more information, please contact:
Ken d’Entremont, Chief Executive Officer Medexus Pharmaceuticals Inc. Tel.: 905-676-0003 E-mail: ken.dentremont@medexus.com
Roland Boivin, Chief Financial Officer Medexus Pharmaceuticals Inc. Tel.: 514-762-2626 ext. 202 E-mail: roland.boivin@medexus.com
Neither the TSX Venture Exchange nor it’s Regulation Services Provider (as that term is defined in the policies of the TSX Venture Exchange) accept responsibility for the adequacy or accuracy of this release.
READER ADVISORIES
Forward-Looking Statements
This press release contains “forward-looking information” within the meaning of applicable securities legislation. Forward-looking information includes, but is not limited to, statements with respect to future business operation, including with respect to the expected growth of the Company’s pharmaceutical portfolio and pipeline. All statements, other than of historical fact, that address activities, events or developments that the Company believes, expects or anticipates will or may occur in the future are forward-looking statements. Forward-looking statements are generally identifiable by use of the words “may”, “will”, “should”, “continue”, “expect”, “anticipate”, “estimate”, “believe”, “intend”, “plan” or “project” or the negative of these words or other variations on these words or comparable terminology. Forward-looking statements are subject to a number of risks and uncertainties, many of which are beyond the Company’s ability to control or predict, that may cause the actual results of the Company to differ materially from those discussed in the forward-looking statements. Factors that could cause actual results or events to differ materially from current expectations include, among other things, without limitation, the risk that the Company will not receive regulatory approvals in a timely manner or at all, the results of certain drug therapies and their impact on the Company’s profitability, the Company’s business plans, and other risks disclosed in the Company’s public disclosure record on file with the relevant securities regulatory authorities. Although Company believes that the expectations and assumptions on which such forward-looking information is based are reasonable, undue reliance should not be placed on the forward-looking information because Company can give no assurance that they will prove to be correct. Since forward-looking information addresses future events and conditions, by its very nature they involve inherent risks and uncertainties. The Company’s actual results, performance or achievement could differ materially from those expressed in, or implied by, the forward-looking information and, accordingly, no assurance can be given that any of the events anticipated by the forward-looking information will transpire or occur, or if any of them do so, what benefits that Company will derive therefrom. Management has included the above summary of assumptions and risks related to forward-looking information provided in this press release in order to provide security holders with a more complete perspective on the Company’s future operations and such information may not be appropriate for other purposes. Readers should not place undue reliance on forward-looking statements. Readers are cautioned that the foregoing lists of factors are not exhaustive. Additional information on these and other factors that could affect the Company’s operations or financial results are included in reports on file with applicable securities regulatory authorities and may be accessed through the SEDAR website (www.sedar.com). The forward-looking statements included in this news release are made as of the date of this news release and the Company does not undertake any obligation to publicly update such forward-looking statements to reflect new information, subsequent events or otherwise unless required by applicable securities legislation.
Plans to submit U.S. and European marketing applications for trilaciclib following regulatory feedback
First clinical data on oral SERD G1T48 expected in 3Q19
Management to host webcast and conference call today at 4:30 p.m. ET
RESEARCH TRIANGLE PARK, N.C., May 09, 2019 (GLOBE NEWSWIRE) — G1 Therapeutics, Inc. (Nasdaq: GTHX), a clinical-stage oncology company, today provided a corporate and financial update for the first quarter ended March 31, 2019.
“Following meetings with regulatory authorities, we have clarity on a path to submitting marketing applications in the U.S. and Europe for trilaciclib based on existing data from our three trials in small cell lung cancer patients,” said Mark Velleca, M.D., Ph.D., Chief Executive Officer. “Our goal is to make trilaciclib available to patients across the globe as quickly as possible and we are encouraged with regulators’ understanding of this new approach to protecting patients from the damaging effects of chemotherapy.”
Raj Malik, M.D., Chief Medical Officer added, “Trilaciclib is the first in a deep pipeline of clinical-stage programs with near-term data readouts expected. We anticipate presenting new data on our next two programs – lerociclib and G1T48 – later this year. Based on promising early results in our Phase 1 clinical trial of G1T48 in ER+ breast cancer, we plan to present proof-of-concept data in the third quarter.”
Clinical, Operational and Executive Team Updates
Plan to submit U.S. and European regulatory filings for trilaciclib: The company announced plans to submit marketing applications in the U.S. and Europe for trilaciclib for myelopreservation in small cell lung cancer (SCLC) based on written feedback from its end-of-Phase 2 meeting with the U.S. Food and Drug Administration (FDA) and discussions with European regulatory authorities. G1 intends to file a New Drug Application (NDA) with the FDA in 2020 and submit a Marketing Authorization Application (MAA) to the European Medicines Agency (EMA) subsequent to an NDA filing. Full press release available here.
Executive team change: Jennifer Moses, who has been with the company for four years and most recently served as Vice President, Finance, has been appointed Chief Financial Officer. Barclay “Buck” Phillips, who had served as CFO and Senior Vice President, Corporate Development since 2017, departed the company to pursue other interests and opportunities.
First investor day presentation: The G1 management team provided a comprehensive overview of the company’s three clinical development programs and outlined the commercialization strategy for trilaciclib. External experts Jeffrey Crawford, M.D., Co-director, Solid Tumor Therapeutics Program, Duke Cancer Institute, and Lowell Hart, M.D., Scientific Director of Research, Florida Cancer Specialists and trilaciclib clinical trial investigator, discussed chemotherapy-induced myelosuppression and trilaciclib’s potential to protect the bone marrow from damage by chemotherapy and improve patient outcomes. The webcast is available on the G1 website here.
First Quarter 2019 Financial Highlights
Cash Position: Cash, cash equivalents and short-term investments totaled $347.8 million as of March 31, 2019, compared to $369.3 million as of December 31, 2018.
Operating Expenses: Operating expenses were $25.9 million for the first quarter of 2019, compared to $20.7 million for the first quarter of 2018. GAAP operating expenses include stock-based compensation expense of $3.8 million for the first quarter of 2019, compared to $1.6 million for the first quarter of 2018.
Research and Development Expenses: Research and development (R&D) expenses for the first quarter of 2019 were $18.1 million, compared to $17.3 million for the first quarter of 2018. The increase in expense was primarily due to an increase in clinical program costs and personnel costs due to additional headcount.
General and Administrative Expenses: General and administrative (G&A) expenses for the first quarter of 2019 were $7.8 million, compared to $3.4 million for the first quarter of 2018. The increase in expense was largely due to an increase in compensation due to headcount increase, increase in pre-commercialization activities and an increase in professional fees and other administrative costs necessary to support our operations as a public company.
Net Loss: G1 reported a net loss of $24.0 million for the first quarter of 2019, compared to $20.4 million for the first quarter of 2018.
Anticipated Milestones for 2019
Expect to complete pre-NDA meeting with the FDA.
Report additional data from all four randomized Phase 2 trilaciclib clinical trials.
Present proof-of-concept data from the Phase 1 clinical trial of G1T48, an oral selective estrogen receptor degrader (SERD), in ER+ breast cancer in Q3 2019.
Present preliminary dose-escalation data from the Phase 1b clinical trial of lerociclib/Tagrisso® (osimertinib) in non-small cell lung cancer in Q3 2019.
Present additional data from the Phase 1b clinical trial of lerociclib/Faslodex® (fulvestrant) in ER+, HER2- breast cancer in Q4 2019.
Webcast and Conference Call The management team will host a webcast and conference call at 4:30 p.m. ET today to provide a corporate and financial update for the first quarter of 2019 ended March 31, 2019. The live call may be accessed by dialing 866-763-6020 (domestic) or 210-874-7713 (international) and entering the conference code:7988598. A live and archived webcast will be available on the Events & Presentations page of the company’s website: www.g1therapeutics.com. The webcast will be archived on the same page for 90 days following the event.
About G1 Therapeutics G1 Therapeutics, Inc. is a clinical-stage biopharmaceutical company focused on the discovery, development, and delivery of innovative therapies that improve the lives of those affected by cancer. The company is advancing three clinical-stage programs. Trilaciclib and lerociclib are designed to enable more effective combination treatment strategies and improve patient outcomes across multiple oncology indications. G1T48 is a potential best-in-class oral selective estrogen receptor degrader (SERD) for the treatment of ER+ breast cancer. G1 also has an active discovery program focused on cyclin-dependent kinase targets.
G1 is based in Research Triangle Park, N.C. For additional information, please visit www.g1therapeutics.com and follow us on Twitter @G1Therapeutics.
Forward-Looking Statements This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “may,” “will,” “expect,” “plan,” “anticipate,” “estimate,” “intend” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. Forward-looking statements in this news release include, but are not limited to, the therapeutic potential of trilaciclib, lerociclib and G1T48 and the timing for next steps with regard to the trilaciclib marketing applications, and are based on the Company’s expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Factors that may cause the Company’s actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in the Company’s filings with the U.S. Securities and Exchange Commission, including the “Risk Factors” sections contained therein and include, but are not limited to, the Company’s ability to complete clinical trials for, obtain approvals for and commercialize any of its product candidates; the Company’s initial success in ongoing clinical trials may not be indicative of results obtained when these trials are completed or in later stage trials; the inherent uncertainties associated with developing new products or technologies and operating as a development-stage company; the Company’s development of a CDK4/6 inhibitor to reduce chemotherapy-induced myelosuppression is novel, unproven and rapidly evolving and may never lead to a marketable product; and market conditions. Except as required by law, the Company assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
This work compares mpMRI with high-resolution 29MHz micro-ultrasound imaging, which maintains the workflow, simplicity and low cost of ultrasound and can be used to target biopsies without the need for MRI.
Giovanni Lughezzani (1), Ander Astobieta (2), Frédéric Staerman (3), Eric Klein (4), Robert Abouassaly (4), Ahmed El-Shefai (4), Gregg Eure (5) (1) Instituto Clinico Humanitas, Rozzano, Italy, (2) Urología Clínica, Clínica IMQ Zorrotzaurre, Spain, (3) Polyclinique Reims-Bezannes, Reims, France, (4) Glickman Urological Institute, Cleveland Clinic, Cleveland, USA, (5) Urology of Virginia, Virginia Beach, USA
Introduction & Objectives
Multi-parametric MRI (mpMRI) is growing as a screening approach for prostate cancer due to high sensitivity and negative predictive value. Unfortunately, implementation is complicated due to additional costs, complexity, learning curve, procedure time, and experience required for adequate interpretation and reproducible results. Many men are also contraindicated due to renal function, claustrophobia, or ferromagnetic implants.
Materials & Methods
280 subjects underwent prostate biopsy using the ExactVu™ micro-ultrasound system (Exact Imaging, Markham, Canada) at 5 urological centers in the USA and Europe
Samples in all subjects were taken from:
mpMRI targets (PI-RADS™ > 2, Cognitive fusion at 4 sites, software-assisted fusion at 1 site)
Micro-ultrasound targets (PRI-MUS™, > 2)
Up to 12 additional systematic samples filled in
Clinically significant cancer was considered any Gleason Sum > 6
Results
Micro-ultrasound provided superior sensitivity to mpMRI for detection of csPCa (p=0.002).
mpMRI demonstrated strong sensitivity (83%), with slightly weaker NPV (69%). Micro-ultrasound sensitivity (95%) and NPV (82%) were both higher. However, micro-ultrasound was less specific (17% vs 25% for mpMRI) and both modalities showed a relatively poor PPV of 43 and 44%.
Conclusions
Micro-ultrasound’s high sensitivity makes it an attractive option for guiding targeted biopsy relative to the more widely studied mpMRI
The slightly lower specificity suggests micro-ultrasound is not yet able to exclude biopsy in as many men, though this is expected to improve as more information on benign variants becomes available
References
Ghai S, Eure G, Fradet V, et al: Assessing Cancer Risk on Novel 29 MHz Micro-Ultrasound Images of the Prostate: Creation of the Micro-Ultrasound Protocol for Prostate Risk Identification. J. Urol. 2016; 196: 562–569.
Astobieta et al. Initial results comparing 29 MHz micro-ultrasound with multi-parametric MRI for targeted prostate biopsy: Relative sensitivity to clinically significant prostate cancer. Eur Urol Suppl. 2018;17(2):e901.
Eure, et al. Comparison of Conventional TRUS, MRI and Micro-Ultrasound for Visualizing Prostate Cancer in an Active Surveillance Population. Can Urol Assoc J. 2018 Aug 30
Lughezzani et al. Comparison between the diagnostic accuracy of high resolution micro-ultrasound versus multiparametric MRI in the detection of prostate cancer: Preliminary results from a single-institutional ongoing prospective trial. Eur Urol Suppl. 2018;17(8):179.
Abouassaly, et al. Initial Results Comparing Micro-Ultrasound with MRI for Prostate Cancer Detection. ESUI 2018
Staerman. Initial Clinical Experience with 29 MHz Micro-Ultrasound for Real-Time Targeted Prostate Biopsies. ESUI 2018
About Exact Imaging
Exact Imagingis the world’s leader in high-resolution micro-ultrasound systems enabling real-time imaging and guided biopsies in the urological market for prostate cancer. Exact Imaging’s ExactVu™ micro-ultrasound platform operates at 29 MHz and enables a whole new level of resolution with the benefits of ease of use, affordability, and is an extension of the current urological workflow. Using the Exact Imaging platform, urologists are able to visualize areas of interest in the prostate and specifically target biopsies at those areas. For the minority of cases where MRI might assist (i.e., prior negative biopsies), the FusionVu™ micro-US/MRI fusion application operates on the ExactVu micro-ultrasound platform and facilitates MRI fusion-based targeting. The ExactVu micro-ultrasound system including the FusionVu application have received regulatory approval in the European Union (CE Mark), the United States (FDA 510(k)) and Canada (Health Canada medical device license).
8 patients with predictable generalized or focal epileptic seizures treated with Staccato alprazolam
62.5% of patients respond with the cessation of seizure activity within two minutes of therapy administration and no recurrence of seizure activity within two hours
Feasibility of both self-administration or caregiver-administration demonstrated
Enrollment of additional 115 patients in double-blind, placebo-controlled part 2 of the study underway
SUMMIT, N.J., May 6, 2019 /PRNewswire/ — Engage Therapeutics, Inc., a clinical stage biopharmaceutical company developing a Rapid Epileptic Seizure Termination (REST) therapy for people with epilepsy who experience a predictable pattern of seizures presented data from part 1 of its Phase 2b StATES Study (Staccato Alprazolam Terminates Epileptic Seizures) at the 2019 American Academy of Neurology (AAN) Annual Meeting on Sunday in Philadelphia.
Eight adult patients with either generalized or focal predictable seizures were enrolled and treated with a single dose of Staccato alprazolam, an investigational therapy that combines FDA-approved Staccato delivery technology with alprazolam, an FDA approved benzodiazepine, in the open-label, run-in portion of this two-part study. Outcome measures were safety, efficacy, pharmacokinetics, and feasibility of self- or staff-caregiver-administration. Highlights from the AAN 2019 poster, entitled, “A Two-Part, Double-Blind, Placebo-Controlled, Inpatient, Dose-Ranging Efficacy Study of Staccato Alprazolam (STAP-001) in Patients with Epilepsy with a Predictable Seizure Pattern: Results from the Initial Open-Label Feasibility Part,” include:
Five of eight (5/8) or 62.5% of patients responded with the cessation of seizure activity within two minutes of treatment administration and no recurrence of seizure activity within two hours;
Responses were observed irrespective of seizure type, BMI or concurrent use of CYP450 inducers;
No serious or severe adverse events reported;
Study procedures for the administration of 1 mg Staccato alprazolam to patients with predictable seizures were deemed feasible;
The double-blind, part 2 portion of the study has initiated and will randomize 115 patients to one of two active arms (1 mg or 2 mg Staccato alprazolam) vs placebo.
“These encouraging data not only illustrate a potential efficacy signal for Staccato alprazolam in rapidly arresting seizures in patients with epilepsy but also support the safety and feasibility of administering this unique therapy to patients just as they begin to experience seizure activity,” said Jaqueline French, MD, the study’s principal investigator and professor in the Department of Neurology at NYU Langone Health’s Comprehensive Epilepsy Center, and founder/director of the Epilepsy Study Consortium. “Currently available seizure therapies are limited to preventing additional seizures, leaving patients with epilepsy in constant fear. A rescue therapy capable of rapidly aborting a seizure could alleviate that fear in many people.”
About the StATES Study The multi-center StATES Study (Staccato Alprazolam Terminates Epileptic Seizures -NCT03478982) is a two-part phase 2b trial designed to evaluate the safety, efficacy, and usability of Staccato alprazolam in subjects with epilepsy who have a predictable seizure pattern. In addition to the eight patients enrolled in the open-label, part 1 portion of the study, up to 115 patients will be enrolled in part 2 of the study, double-blinded and randomized to one of two active arms (1 mg or 2 mg Staccato Alprazolam) vs placebo. The primary endpoint of the study is cessation of seizure activity within two minutes of treatment administration and no recurrence within two hours based on clinical observation. Additional endpoints include safety and tolerability, the severity of the seizures compared to a subject’s prior seizures, utilization of rescue medication and incidence of adverse events. The StATES study is being conducted at approximately 50 trial sites in the United States, Australia, Canada, and Jamaica. Please visit www.epilepsyhealthstudy.com for more information about the StATES study and www.engagetherapeutics.com/study-locations/ for a list of study locations.
About Staccato Alprazolam
Staccato alprazolam is a single-use, investigational epileptic seizure rescue therapy that combines the Staccato delivery technology, which is currently used in a U.S. Food and Drug Administration (FDA) approved product, with alprazolam, an FDA-approved benzodiazepine. It is a small, easy-to-use, hand-held inhaler that delivers alprazolam with a single breath potentially providing a way for people with epilepsy and their caregivers to stop an ongoing seizure. The Staccato system rapidly vaporizes alprazolam to form an aerosol, with particle size designed for deep lung delivery, producing a rapid, systemic effect. In a phase 2a proof-of-concept study, Staccato alprazolam demonstrated rapid reduction of seizure-like activity in a photosensitivity model.
About Engage Therapeutics, Inc.
Engage Therapeutics is developing Staccato alprazolam for the immediate cessation of active and acute epileptic seizures. The investigational product is in the Rapid Epileptic Seizure Termination (REST) category of products. Engage Therapeutics is based in Summit, N.J.
VANCOUVER, Canada–(BUSINESS WIRE)– Zymeworks Inc. (NYSE/TSX: ZYME), a clinical-stage biopharmaceutical company developing multifunctional biotherapeutics, is pleased to announce the detailed voting results on the items of business considered at its Annual General Meeting of Shareholders held on May 2, 2019 (the “Meeting”).
Shareholder Voting Results
The Shareholders voted on the following matters at this year’s Meeting.
Proposal 1 – Election of Directors
The nominees listed in Zymeworks’ proxy statement dated March 18, 2019 (the “Proxy Statement”) were elected as Directors of the Company. Detailed results of the votes are set out below:
Proposal 1
Outcome of the Vote
Votes by Ballot
Election of Directors
Votes For
Votes Withheld
Kenneth Hillan
Carried
18,394,190 (99.92%)
14,339 (0.08%)
Natalie Sacks
Carried
18,395,416 (99.93%)
13,113 (0.07%)
Proposal 2 – Appointment of Auditors
The vote was carried for the Appointment of the Auditors, KPMG LLP. Detailed results of the votes are set out below:
Proposal 2
Outcome of the Vote
Votes by Ballot
Votes For
Votes Withheld
Appointment of KPMG LLP
Carried
23,430,739 (99.88%)
28,799 (0.12%)
Full details of all proposals are fully described in the Proxy Statement available on the Company’s profile on SEDAR and on EDGAR and the detailed results of voting on each proposal are included in the Report of Voting Results filed on SEDAR and on EDGAR.
About Zymeworks Inc.
Zymeworks is a clinical-stage biopharmaceutical company dedicated to the development of next-generation multifunctional biotherapeutics. The Company’s suite of therapeutic platforms and its fully integrated drug development engine enable precise engineering of highly differentiated product candidates. Zymeworks’ lead clinical candidate, ZW25, is a novel AzymetricTM bispecific antibody currently in Phase 2 clinical development. The Company’s second clinical candidate, ZW49, is a bispecific antibody-drug conjugate currently in Phase 1 clinical development and combines the unique design and antibody framework of ZW25 with Zymeworks’ proprietary ZymeLinkTM cytotoxic payload. Zymeworks is also advancing a deep preclinical pipeline in immuno-oncology and other therapeutic areas. In addition, its therapeutic platforms are being leveraged through multiple strategic partnerships with eight global biopharmaceutical companies.
Zymeworks Inc. Investor Inquiries: Ryan Dercho, Ph.D. (604) 678-1388 ir@zymeworks.com
MONTREAL, May 7, 2019 — Medexus Pharmaceuticals Inc. (the “Company” or “Medexus”) (TSXV: MDP, OTCQB: PDDPF), today announced the launch of a new Metoject® Subcutaneous 15mg dose in Canada. Metoject Subcutaneous is a pre-filled syringe of methotrexate with a pre-attached subcutaneous needle that is approved in Canada for the treatment of rheumatoid arthritis, psoriasis, and psoriatic arthritis.
Metoject Subcutaneous 15mg is currently reimbursed by the Provinces of Saskatchewan, New Brunswick, Nova Scotia, and Newfoundland and Labrador through their provincial formularies, as well as the Yukon Non-Insured Health Benefits (NIHB) and Correctional Services Canada. As of May 1, 2019, Metoject Subcutaneous 15 mg is reimbursed in the Province of Ontario by the Ontario Drug Benefit Program (ODB). Metoject Subcutaneous 15 mg dose is in addition to the dosage strengths of 17.5mg, 20mg, 22.5mg, and 25mg currently offered in Canada by Medexus.
Ken d’Entremont, Chief Executive Officer of Medexus, commented, “The new 15mg dose of Metoject Subcutaneous is an important addition to the Metoject line in Canada, as it gives physicians more flexibility to accurately prescribe an appropriate strength for their patients, and offers patients a more convenient and comfortable option. We expect this dose to be a significant portion of our Metoject volume going forward.”
About Medexus
Medexus is a leading specialty pharmaceutical company with a strong North American commercial platform. The Company’s vision is to provide the best healthcare products to healthcare professionals and patients, through our core values of Quality, Innovation, Customer Service and Teamwork. Medexus Pharmaceuticals is focused on the therapeutic areas of auto-immune disease and pediatrics. The leading products are Rasuvo and Metoject, a unique formulation of methotrexate (auto-pen and pre-filled syringe) designed to treat rheumatoid arthritis and other auto-immune diseases; and Rupall, an innovative prescription allergy medication with a unique mode of action.
For more information, please contact:
Ken d’Entremont, Chief Executive Officer Medexus Pharmaceuticals Inc. Tel.: 905-676-0003 E-mail: ken.dentremont@medexus.com
Roland Boivin, Chief Financial Officer Medexus Pharmaceuticals Inc. Tel.: 514-762-2626 ext. 202 E-mail: roland.boivin@medexus.com
Neither the TSX Venture Exchange nor its Regulation Services Provider (as that term is defined in the policies of the TSX Venture Exchange) accepts responsibility for the adequacy or accuracy of this release.
READER ADVISORIES
Forward-Looking Statements
This press release contains “forward-looking information” within the meaning of applicable securities legislation. Forward-looking information includes, but is not limited to, statements with respect to future business operation, including with respect to the expected growth of the Company’s pharmaceutical portfolio and pipeline. All statements, other than of historical fact, that address activities, events or developments that the Company believes, expects or anticipates will or may occur in the future are forward-looking statements. Forward-looking statements are generally identifiable by use of the words “may”, “will”, “should”, “continue”, “expect”, “anticipate”, “estimate”, “believe”, “intend”, “plan” or “project” or the negative of these words or other variations on these words or comparable terminology. Forward-looking statements are subject to a number of risks and uncertainties, many of which are beyond the Company’s ability to control or predict, that may cause the actual results of the Company to differ materially from those discussed in the forward-looking statements. Factors that could cause actual results or events to differ materially from current expectations include, among other things, without limitation, the risk that the Company will not receive regulatory approvals in a timely manner or at all, the results of certain drug therapies and their impact on the Company’s profitability, the Company’s business plans, and other risks disclosed in the Company’s public disclosure record on file with the relevant securities regulatory authorities. Although Company believes that the expectations and assumptions on which such forward-looking information is based are reasonable, undue reliance should not be placed on the forward-looking information because Company can give no assurance that they will prove to be correct. Since forward-looking information addresses future events and conditions, by its very nature they involve inherent risks and uncertainties. The Company’s actual results, performance or achievement could differ materially from those expressed in, or implied by, the forward-looking information and, accordingly, no assurance can be given that any of the events anticipated by the forward-looking information will transpire or occur, or if any of them do so, what benefits that Company will derive therefrom. Management has included the above summary of assumptions and risks related to forward-looking information provided in this press release in order to provide securityholders with a more complete perspective on the Company’s future operations and such information may not be appropriate for other purposes. Readers should not place undue reliance on forward-looking statements. Readers are cautioned that the foregoing lists of factors are not exhaustive. Additional information on these and other factors that could affect the Company’s operations or financial results are included in reports on file with applicable securities regulatory authorities and may be accessed through the SEDAR website (www.sedar.com). The forward-looking statements included in this news release are made as of the date of this news release and the Company does not undertake an obligation to publicly update such forward-looking statements to reflect new information, subsequent events or otherwise unless required by applicable securities legislation.
SEATTLE, May 7, 2019 /PRNewswire/ — Bardy Diagnostics, Inc., (“BardyDx”), a leading provider of ambulatory cardiac monitoring technologies and custom data solutions, announced it has been selected as the winner of the North American Remote Cardiac Monitoring Technology Innovation Award by Frost & Sullivan, a leader in global research and consulting solutions. BardyDx earned the distinction for its industry-leading advancements in remote cardiac monitoring led by its flagship product, the Carnation Ambulatory Monitor (“CAM™”), the only P-wave centric™ ambulatory cardiac patch monitor and arrhythmia detection device.
“Recognition of the CAM™ patch and our P-wave centric™ platform as a breakthrough in ECG monitoring is a direct reflection of our team’s dedication to developing and marketing inventive solutions that reveal the true message and meaning of a patient’s cardiac rhythm, messages that are frequently ignored or missed using existing technologies,” said Gust H. Bardy, MD, Founder and Chief Executive Officer of Bardy Diagnostics. “This prestigious honor recognizes our experience and our years of clinical and technical research, development, and innovation.”
Frost & Sullivan specifically praised the CAM™ patch’s pioneering advancements in engineering and design that have quickly become the industry standard for high fidelity ECG datasets including accurate and clear P-wave detection and recording. BardyDx was also recognized for its advances in artificial intelligence (AI)’s assistive role in enhancing human capabilities for analyzing and interpreting heart rhythms made possible, in part, by the CAM™ patch’s ECG signal quality. Frost & Sullivan also reported that the evolution towards BardyDx’s “augmented” intelligence is “achievable solely because of the CAM™ patch’s unique ability to accurately detect a broad array of arrhythmias, including those commonly mistaken for atrial fibrillation by commercially-available, R-wave focused automated detectors.”
Frost & Sullivan Best Practices Awards recognize companies in a variety of regional and global markets for demonstrating outstanding achievement and superior performance in areas such as leadership, technological innovation, customer service, and strategic product development. Industry analysts compare market participants and measure performance through in-depth interviews, analysis, and extensive secondary research to identify best practices in the industry.
“Overall, BardyDx’s thought leadership, unparalleled innovation, and disruptive potential have positioned it for greater adoption in the cardiac monitoring market,” said Kamaljit Behera, Industry Analyst at Frost & Sullivan.
This award builds upon the growing market recognition of the innovative P-wave centric CAM™ patch. Most recently, BardyDx was named the winner of the 2019 GeekWire Award in the Hardware of the Year category presented at the 11th annual GeekWire Awards Ceremony held May 2, 2019. Hosted by GeekWire, a Seattle-based technology news site that provides national breaking news, expert analysis and unique insights into the evolving technology industry to a worldwide tech-savvy audience, the Awards honor stars of the Pacific Northwest startup and tech community each year. Also, BardyDx was recently named the winner of the Impact Pediatric Health Competition hosted by the nation’s leading pediatric healthcare institutions at SXSW 2019 for the CAM™’s unique pediatric-friendly design and potential to address significant unmet needs in pediatric healthcare. In addition, BardyDx was selected as the winner of the 2018 Fierce Innovation Life Sciences Award for Medical Device Innovation from the leading industry publisher of FierceBiotech & FiercePharma.
40th Annual Heart Rhythm Scientific Sessions
BardyDx will be exhibiting at the 40th Annual Heart Rhythm Scientific Sessions in San Francisco on May 8-10, 2019. Visit booth #2243 to learn about the CAM™ patch and the innovative P-wave centric platform.
About Frost & Sullivan:
Frost & Sullivan, the Growth Partnership Company, works in collaboration with clients to leverage visionary innovation that addresses the global challenges and related growth opportunities that will make or break today’s market participants. For more than 50 years, we have been developing growth strategies for the global 1000, emerging businesses, the public sector, and the investment community.
About Bardy Diagnostics:
Bardy Diagnostics, Inc. is an innovator in digital health and remote patient monitoring, with a focus on providing diagnostically-accurate and patient-friendly cardiac patch and other monitors to the industry. The company’s CAM™ patch is a non-invasive, P-wave centric™ ambulatory cardiac monitor and arrhythmia detection device that is designed to improve patient compliance for adults and children through its lifestyle-enabling design. Designed to be worn comfortably and discreetly, the female-friendly, hourglass-shaped CAM™ patch is placed on the center of the chest, directly over the heart for optimum ECG signal collection. The proprietary technology of the CAM™ patch provides optimal detection and clear recording of the often difficult-to-detect P-wave, the signal of the ECG waveform that is essential for accurate arrhythmia diagnosis.
MEDIA CONTACT: Jonathan Wu Director, Marketing Bardy Diagnostics, Inc. 1-844-422-7393 jwu@bardydx.com
New EV5C Abdominal Transducer and Color Flow and Power Doppler capabilities enable the ExactVu™ micro-ultrasound system to provide a complete men’s health urological imaging solution
TORONTO, CANADA — (May 7, 2019) Exact Imaging, the world’s leader in high-resolution micro-ultrasound systems enabling real-time imaging and biopsy guidance for the prostate, announced it has received CE Mark Approval for its Abdominal Imaging Release including its new EV5C Abdominal Transducer and Color Flow and Power Doppler capabilities. Operating on the ExactVu™ micro-ultrasound system, the EV5C Abdominal Transducer and Color Flow and Power Doppler capabilities allow urologists to perform complete urological ultrasound assessments including kidney examinations and biopsies, male pelvic and bladder examinations. The ExactVu™ system, which is the only urological ultrasound platform to operate both conventional as well as high resolution (29 MHz) transducers, now provides the ultimate “total men’s health” imaging solution. It can provide a complete abdominal urological ultrasound assessment of a patient as well as providing the fastest, simplest real-time targeting of prostate biopsies, while providing the highest real-time resolution for guidance for those prostate biopsies.
“We are thrilled to have received our CE mark approval for our Abdominal Imaging Release. The demand is significant for a urological imaging platform that merges the 70-micron resolution of the ExactVu™ micro-ultrasound system for real-time targeted prostate biopsies with the ability to provide a complete urological ultrasound assessment of a patient,” said Randy AuCoin, Exact Imaging’s President and CEO. “The goal is to provide urologists with the most flexible, innovative imaging solution to help urologists better diagnose urological disease. We believe this release helps to further establish the ExactVu™ platform as a most useful, cost-effective men’s health imaging solution.”
About Exact Imaging
Exact Imaging is the world’s leader in high-resolution micro-ultrasound systems enabling real-time imaging and guided biopsies in the urological market for prostate cancer. Exact Imaging’s ExactVu™ micro-ultrasound platform operates at 29 MHz and enables a whole new level of resolution with the benefits of ease of use, affordability, and is an extension of the current urological workflow. Using the Exact Imaging platform, urologists are able to visualize areas of interest in the prostate and specifically target biopsies at those areas. For those cases where MRI might assist, the FusionVu™ micro-US/MRI fusion application operates on the ExactVu™ micro-ultrasound platform and facilitates fast, simple MRI fusion-based targeting with the guidance of the micro-ultrasound system’s 70-micron real-time resolution. The ExactVu™ micro-ultrasound system including the FusionVu application have received regulatory approval in the European Union (CE Mark), the United States (FDA 510(k)) and Canada (Health Canada medical device license). Regulatory approval for the EV5C Abdominal transducer and Color Flow and Power Doppler capabilities in the United States and Canada is pending.
VICTORIA, British Columbia–(BUSINESS WIRE) — Aurinia Pharmaceuticals Inc. (NASDAQ:AUPH / TSX:AUP) (“Aurinia” or the “Company”), a clinical stage biopharmaceutical company focused on the global immunology market, today announced it has appointed Dr. Daniel Billen to its Board of Directors. Dr. Billen’s appointment is effective April 29, 2019 concurrent with the previously announced appointment of Mr. Peter Greenleaf as Chief Executive Officer and elevation of Dr. George Milne to Chairman of the Board.
“On behalf of the entire organization, it is a pleasure to welcome Daniel onto Aurinia’s Board of Directors. Throughout his career, Daniel has led the international growth of novel biopharmaceutical treatments for patients with debilitating kidney and inflammatory diseases, and we look forward to his strategic insight and guidance with the ongoing development of voclosporin,” commented Dr. George Milne, Chairman of the Board of Directors.
Dr. Billen has more than four decades of experience leading the commercialization of pharmaceutical and biotech products in North America and Europe. Prior to his retirement, Dr. Billen served as Vice President and General Manager, Inflammation and Nephrology at Amgen, from 2011 until 2018. Prior to that, Dr. Billen was General Manager, Amgen Canada, from 1991 until 2011. Dr. Billen previously served in roles of escalating responsibility at Janssen from 1979 until 1991. Dr. Billen received his Ph.D. in Chemistry from the University of Louvain, Belgium.
“Voclosporin represents a potentially significant advancement for patients with life-threatening kidney diseases, such as lupus nephritis,” commented Dr. Billen. “After years of admiring Aurinia’s development of voclosporin, it is an honour to now be joining the Board of Directors and helping to guide this exciting program toward commercialization.”
About Aurinia
Aurinia Pharmaceuticals is a late clinical-stage biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are impacted by serious diseases with a high unmet medical need. The Company is currently developing voclosporin, an investigational drug, for the potential treatment of LN, FSGS, and DES. The Company is headquartered in Victoria, British Columbia and focuses its development efforts globally.
About Voclosporin
Voclosporin, an investigational drug, is a novel and potentially best-in-class calcineurin inhibitor (“CNI”) with clinical data in over 2,400 patients across indications. Voclosporin is an immunosuppressant, with a synergistic and dual mechanism of action. By inhibiting calcineurin, voclosporin blocks IL-2 expression and T-cell mediated immune responses and stabilizes the podocyte in the kidney. It has been shown to have a more predictable pharmacokinetic and pharmacodynamic relationship (potentially requires no therapeutic drug monitoring), an increase in potency (vs cyclosporin), and an improved metabolic profile compared to legacy CNIs. Aurinia anticipates that upon regulatory approval, patent protection for voclosporin will be extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act and comparable laws in other countries and until April 2028 with anticipated pediatric extension. Further, the new Notice of Allowanceis expected to result in the issuance of a U.S. patent with a term extending to December 2037. If the FDA approves the use of voclosporin for LN and the label for such use follows the dosing protocol under the Notice of Allowance, the issuance of this patent will expand the scope of intellectual property protection for voclosporin to December 2037.
Forward-Looking Statements
Certain statements made in this press release may constitute forward-looking information within the meaning of applicable Canadian securities law and forward-looking statements within the meaning of applicable United States securities law. These forward-looking statements or information include but are not limited to statements or information with respect to: voclosporin being a potentially best-in-class CNI; patent protection for voclosporin being extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 and until April 2028 with an anticipated pediatric extension; and intellectual property protection for voclosporin being extended to December 2037 in respect of a patent anticipated to be issued in connection with a new Notice of Allowance. It is possible that such results or conclusions may change based on further analyses of these data. Words such as “anticipate”, “will”, “believe”, “estimate”, “expect”, “intend”, “target”, “plan”, “goals”, “objectives”, “may” and other similar words and expressions, identify forward-looking statements. We have made numerous assumptions about the forward-looking statements and information contained herein, including among other things, assumptions about: the costs and expenses associated with Aurinia’s clinical trials; Aurinia receiving approval from regulators to proceed with commercialization; Aurinia being able to complete its clinical trials in a timely fashion; Aurinia being able to extend its patents on terms acceptable to Aurinia; and the validity of our patents. Even though the management of Aurinia believes that the assumptions made, and the expectations represented by such statements or information are reasonable, there can be no assurance that the forward-looking information will prove to be accurate.
Forward-looking information by their nature are based on assumptions and involve known and unknown risks, uncertainties and other factors which may cause the actual results, performance or achievements of Aurinia to be materially different from any future results, performance or achievements expressed or implied by such forward-looking information. Should one or more of these risks and uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those described in forward-looking statements or information. Such risks, uncertainties and other factors include, among others, the following: difficulties, delays, or failures we may experience in the conduct of our AURORA clinical trial; difficulties we may experience in completing the development and commercialization of voclosporin; the market for the LN business may not be as estimated; and regulatory authorities not granting approval for use of voclosporin in a commercial manner, or not granting patents or extensions for patents at all or as Aurinia currently anticipates. Although we have attempted to identify factors that would cause actual actions, events or results to differ materially from those described in forward-looking statements and information, there may be other factors that cause actual results, performances, achievements or events to not be as anticipated, estimated or intended. Also, many of the factors are beyond our control. There can be no assurance that forward-looking statements or information will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, you should not place undue reliance on forward-looking statements or information.
Except as required by law, Aurinia will not update forward-looking information. All forward-looking information contained in this press release is qualified by this cautionary statement. Additional information related to Aurinia, including a detailed list of the risks and uncertainties affecting Aurinia and its business can be found in Aurinia’s most recent Annual Information Form available by accessing the Canadian Securities Administrators’ System for Electronic Document Analysis and Retrieval (SEDAR) website at www.sedar.com or the U.S. Securities and Exchange Commission’s Electronic Document Gathering and Retrieval System (EDGAR) website at www.sec.gov/edgar.
VANCOUVER, British Columbia–(BUSINESS WIRE) May 2, 2019– Zymeworks Inc. (NYSE/TSX: ZYME), a clinical-stage biopharmaceutical company developing multifunctional therapeutics, today reported financial results for the first quarter ended March 31, 2019.
“We are pleased to have advanced both of our lead assets to the next stages of clinical development; recently commencing a Phase 2 study for ZW25 and starting enrollment in the Phase 1 clinical trial for our antibody-drug conjugate, ZW49,” said Ali Tehrani, Ph.D., Zymeworks’ President & CEO. “Accordingly, we have also exp
anded our leadership team, adding experienced executives with critical competencies needed to facilitate the development and approval of our clinical-stage assets. We believe we are now well-positioned to deliver on our ambitious clinical goals throughout 2019 and beyond.”
First Quarter 2019 Business Highlights and Recent Developments
Phase 2 Clinical Trial Begins for ZW25 in First-Line HER2 Expressing Metastatic Gastroesophageal Cancers
The Phase 2 trial is evaluating ZW25 in combination with standard of
care (SOC) chemotherapy for the first-line treatment of HER2-positive
metastatic gastroesophageal cancers. This trial is intended to support a
potential first-line registrational trial and could position ZW25 as a new SOC.
Phase 1 ZW49 Clinical Study Open and Enrolling Patients
Enrollment is underway in the United States for the Phase
1 clinical trial of ZW49, Zymeworks’ novel bispecific HER2-targeted
antibody-drug conjugate. The objectives of this study are to evaluate
safety and early anti-tumor activity as well as establish a recommended dose
for future clinical trials.
Three Experienced Development Executives Added to Management Team
Zymeworks expanded its leadership team and added key functional
expertise to support the development of its maturing clinical pipeline. The
newly created positions include Neil Josephson, M.D., Vice President,
Clinical Research; Bruce Hart, Ph.D., Vice President, Regulatory Affairs;
and Mark Hollywood, Senior Vice President, Technical and Manufacturing
Operations.
Eli Lilly and Daiichi Sankyo Programs Advance Toward Clinical Testing
Zymeworks’ partner, Eli Lilly, filed an Investigational New Drug
Application for its second Azymetric™ program, triggering a US$8.0 million
payment to Zymeworks. In addition, Daiichi Sankyo recently exercised its option
for a commercial license to an immuno-oncology bispecific built using
Zymeworks’ Azymetric and EFECT™ platforms. Zymeworks will receive a US$3.5
million payment.
Financial Results for the Quarter Ended March 31, 2019
Revenue for the three months ended March 31, 2019 was $11.9 million as compared to $0.04 million in the same period of 2018. Revenue for 2019 includes an $8.0 million development milestone payment upon Lilly’s submission of an investigational new drug application, $3.5 million of recognized deferred revenue from our licensing and collaboration agreement with BeiGene, as well as $0.4 million in other research support payments. Revenue for the same period in 2018 was $0.04 million, consisting of research support payments.
For the three months ended March 31, 2019, research and development expenses were $17.5 million as compared to $13.1 million in the first three months of the prior year. The change was primarily due to an increase in clinical trial activity and associated drug manufacturing costs for ZW25, as well as an increase in other research and discovery activities as compared to the same period in 2018. Research and development expenses also included non-cash stock-based compensation expense of $1.1 million from equity classified equity awards and $0.4 million expense related to the non-cash mark-to-market revaluation of certain historical liability classified equity awards.
For the three months ended March 31, 2019, general and administrative expenses were $9.0 million as compared to $7.1 million in the first quarter of 2018. The change was primarily due to an increase in employee compensation expenses from increased head count in 2019 over 2018, including non-cash stock-based compensation, as well as other increases in professional fees associated with year-over-year corporate growth. General and administrative expenses included non-cash stock-based compensation expense of $1.5 million from equity classified equity awards and $1.3 million expense related to the non-cash mark-to-market revaluation of certain historical liability classified equity awards.
The net loss for the three months ended March 31, 2019, was $13.6 million as compared to $21.2 million in the same period of 2018. This was primarily due to increased revenue, interest income and 2018 warrant valuation expense, which was not relevant for 2019, that offset an increase in research and development expenses associated with our lead therapeutic candidates and other programs as well as general and administrative expenses.
Zymeworks expects research and development expenditures to increase over time in line with the advancement and expansion of the Company’s clinical development of its product candidates, as well as its ongoing preclinical research activities. Additionally, Zymeworks anticipates continuing to receive revenue from its existing and future strategic partnerships, including technology access fees and milestone-based payments. However, Zymeworks’ ability to receive these payments is dependent upon either Zymeworks or its collaborators successfully completing specified research and development activities.
As of March 31, 2019, Zymeworks had $180.3 million in cash and cash equivalents and short-term investments.
About Zymeworks Inc.
Zymeworks is a clinical-stage biopharmaceutical company dedicated to the development of next-generation multifunctional biotherapeutics. The Company’s suite of therapeutic platforms and its fully integrated drug development engine enable precise engineering of highly differentiated product candidates. Zymeworks’ lead clinical candidate, ZW25, is a novel Azymetric™ bispecific antibody currently in Phase 2 clinical development. The Company’s second clinical candidate, ZW49, is a bispecific antibody-drug conjugate currently in Phase 1 clinical development and combines the unique design and antibody framework of ZW25 with Zymeworks’ proprietary ZymeLink™ cytotoxic payload. Zymeworks is also advancing a deep preclinical pipeline in immuno-oncology and other therapeutic areas. In addition, its therapeutic platforms are being leveraged through multiple strategic partnerships with eight global biopharmaceutical companies. For more information, visit www.zymeworks.com.
This press release includes “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. Forward-looking statements in this news release include, but are not limited to, statements that relate to potential payments and/or royalties payable to Zymeworks under its corporate agreements, the speed and outcome of drug development plans, ZW25’s ability to be approved and become a standard-of-care treatment for certain types of cancer, Zymeworks’ potential global growth, and other information that is not historical information. When used herein, words and phrases such as “enable”, “will”, “may”, “expect”, “anticipate”, “eligible to”, and similar expressions are intended to identify forward-looking statements. In addition, any statements or information that refer to expectations, beliefs, plans, projections, objectives, performance or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking. All forward-looking statements are based upon Zymeworks’ current expectations and various assumptions. Zymeworks believes there is a reasonable basis for its expectations and beliefs, but they are inherently uncertain. Zymeworks may not realize its expectations, and its beliefs may not prove correct. Actual results could differ materially from those described or implied by such forward-looking statements as a result of various factors, including, without limitation, market conditions and the factors described under “Risk Factors” in Zymeworks’ Quarterly Report on Form 10-Q for its quarter ended March 31, 2019 (a copy of which may be obtained at www.sec.gov and www.sedar.com). Consequently, forward-looking statements should be regarded solely as Zymeworks’ current plans, estimates and beliefs. Investors should not place undue reliance on forward-looking statements. Zymeworks cannot guarantee future results, events, levels of activity, performance or achievements. Zymeworks does not undertake and specifically declines any obligation to update, republish, or revise any forward-looking statements to reflect new information, future events or circumstances or to reflect the occurrences of unanticipated events, except as may be required by law.
NON-GAAP FINANCIAL MEASURES
In addition to reporting financial information in accordance with U.S. generally accepted accounting principles (“GAAP”) in this press release, Zymeworks is also reporting normalized expenses and normalized loss per share, which are non-GAAP financial measures. Normalized expenses and normalized loss per share are not defined by GAAP and should not be considered as alternatives to net loss, net loss per share or any other indicator of Zymeworks’ performance required to be reported under GAAP. In addition, Zymeworks’ definitions of normalized expenses and normalized loss per share may not be comparable to similarly titled non-GAAP measures presented by other companies. Investors and others are encouraged to review Zymeworks’ financial information in its entirety and not rely on a single financial measure. As defined by Zymeworks, normalized expenses represent total research and development expenses and general and administrative expenses adjusted for non-cash stock-based compensation expenses for equity and liability classified equity instruments.
Normalized expenses are a non-GAAP measure that Zymeworks believes is useful because it excludes those items that Zymeworks believes are not representative of Zymeworks’ operating expenses.
Micro-Ultrasound and its Significant Clinical Benefits are Featured at the Largest Prostate Cancer Conference in 2 Clinical Talks, 5 Oral Presentations and Clinical Posters and a Skills Symposium
CHICAGO, IL — (May 3, 2019) Exact Imaging, the world’s leader in high-resolution micro-ultrasound systems enabling real-time imaging and biopsy guidance for the prostate, and its ExactVu™ micro-ultrasound system will be featured prominently at the 114th Annual Meeting of the AUA (American Urological Association)in Chicago, IL from May 3 – 6, 2019. There will be a podium talk on the “High Resolution 29 MHz Micro-Ultrasound in the Diagnosis of Primary and Recurrent Prostate Cancer” (Session PD23-03) by Drs. Klotz and Woon (Sunnybrook Health Sciences Center, Toronto, CA) and a second podium talk on “A Multi-Institutional Randomized Controlled Trial Comparing Novel First Generation High-Resolution Micro-Ultrasound with Conventional Frequency Ultrasound for Transrectal Prostate Biopsy” (Session PD23-04) by Dr. Christian Pavlovich (Johns Hopkins University, Baltimore, USA), both on Saturday, May 4, 2019. A Skills Enhancement Workshop on “Targeted Prostate Biopsies using Micro-Ultrasound” on Sunday, May 5th will feature the following 3 talks:
ExactVu™ Micro-Ultrasound for Targeted Prostate Biopsies: 2 Years of Clinical Experience and Growing Body of Clinical Evidence (Dr. Gregg Eure, Urology of Virginia, Virginia Beach, VA)
Comparison of Cancer Detection Rate of ExactVu™ Micro-Ultrasound vs. mpMRI/MRI Fusion (Dr. Rafael Sanchez-Salas, L’institut Mutualiste Montsouris (l’IMM), Paris, FR)
Significant Potential of 29 MHz Micro-Ultrasound for Urologic Imaging — and Potential for Personalized Treatment (Dr. Daniel Rukstalis, Wake Forest Baptist Health, Winston-Salem, NC)
In addition, the following 5 posters citing the clinical utility of the ExactVu™ micro-ultrasound system will also be presented at the conference:
Diagnostic Accuracy of Targeted Prostate Biopsies:
Results from a Prospective Trial Comparing Micro-Ultrasound with Multiparametric
MRI for the Detection of Prostate Cancer Davide Maffei
et al, Humanitas Clinical and Research Center, Rozzano,
Italy
Exact Imaging will also be exhibiting the ExactVu™ platform, along with the FusionVu™ application during the commercial exhibition in Booth #2111. The conference and exhibit will be held at the McCormick Place, Chicago, IL.
“The growing number of talks, posters and workshops on the ExactVu™ micro-ultrasound system reflects the utility and growing pervasiveness of micro-ultrasound as a leading tool for urologists worldwide to guide their targeted biopsies,” says Randy AuCoin, Exact Imaging’s President and CEO. “The ExactVu™ micro-ultrasound system provides the highest real-time resolution for targeted prostate biopsies. The results being presented from across our extensive network of customers shows the usefulness of the system for improving practitioners’ abilities to visualize and guide their targeted prostate biopsies and then follow treatment pathways”.
About Exact Imaging:
Exact Imagingis the world’s leader in high-resolution micro-ultrasound systems enabling real-time imaging and guided biopsies in the urological market for prostate cancer. Exact Imaging’s ExactVu™ micro-ultrasound platform operates at 29 MHz and enables a whole new level of resolution with the benefits of ease of use, affordability, and is an extension of the current urological workflow. Using the Exact Imaging platform, urologists are able to visualize areas of interest in the prostate and specifically target biopsies at those areas. For those cases where MRI might assist, the FusionVu™ micro-US/MRI fusion application operates on the ExactVu™ micro-ultrasound platform and facilitates fast, simple MRI fusion-based targeting with the guidance of the micro-ultrasound system’s 70-micron real-time resolution. The ExactVu™ micro-ultrasound system including the FusionVu™ application have received regulatory approval in the European Union (CE Mark), the United States (FDA 510(k)) and Canada (Health Canada medical device license).
QUEBEC CITY, April 30, 2019 /CNW Telbec/ –OpsensInc. (“Opsens” or the “Company”) (TSX: OPS) (OTCQX: OPSSF) is pleased to announce it has entered into a supply agreement as part of its long-term collaboration with Abiomed, Inc. (“Abiomed”) for the Impella CP® heart pump.
Abiomed has awarded Opsens a five-year agreement to supply a critical
component for its heart pump technology. The contract includes mutual
renewal clauses. This new agreement between Opsens and Abiomed follows a
co-development and license agreement to integrate Opsens’ miniature
optical pressure sensor into Impella CP® heart pumps, signed in 2014.
“We are pleased to significantly expand our collaboration with Abiomed and the integration of our optical technology into the widely used Impella CP® in the United States. This partnership clearly highlights the benefits of our optical technology for cardiac applications and demonstrates the accuracy of our measurement technology as well as the quality of our manufacturing capabilities,” said Louis Laflamme, President and CEO of Opsens.
“The continued development of our original equipment
manufacturer (OEM) business segment is strategically important for
Opsens,” adds Laflamme. It increases the critical mass in manufacturing,
further advancing the company’s improvement initiatives. This agreement
allows Opsens to capitalize on the work done with Abiomed in recent
years and highlights the quality of its offer to the interventional
cardiology market globally.
About Opsens Inc.
Opsens focuses mainly on physiological measurements such as FFR and dPR in interventional cardiology. Opsens offers an advanced optical-based pressure guidewire that aims at improving the clinical outcome of patients with coronary artery disease. Its flagship product, the OptoWire, is a second-generation fiber optic pressure guidewire designed to provide the lowest drift in the industry and excellent lesions access. The OptoWire has been used in the diagnosis and treatment of over 60,000 patients in more than 30 countries. It is approved for sale in the United States, European Union, Japan, and Canada.
Opsens is also involved in industrial activities in developing,
manufacturing and installing innovative fibre optic sensing solutions
for critical applications.
Forward-looking statements contained in this press release involve
known and unknown risks, uncertainties and other factors that may cause
actual results, performance and achievements of Opsens to be materially
different from any future results, performance or achievements
expressed or implied by the said forward-looking statements.
Neither TSX nor its Regulation Services Provider (as that term is
defined in the policies of the TSX) accepts responsibility for the
adequacy or accuracy of this release.
For further information:
Louis Laflamme, CPA, CA, Chief Executive Officer, 418.781.0333
Robin Villeneuve, CPA, CA, Chief Financial Officer, 418.781.0333
VANCOUVER, British Columbia / BUSINESSWIRE/ Zymeworks Inc. (NYSE/TSX: ZYME), a clinical-stage biopharmaceutical company developing multifunctional biotherapeutics, today announced it has expanded its leadership team with the hiring of three new vice presidents to support the Company’s expanded global clinical development: Mark Hollywood, Senior Vice President, Technical and Manufacturing Operations; Neil Josephson, M.D., Vice President, Clinical Research; and Bruce Hart, Ph.D., Vice President, Regulatory Affairs. Zymeworks has also promoted David Poon, Ph.D., to Vice President, Business Development and Alliance Management.
“Adding these seasoned professionals to our leadership team further expands the critical expertise required to advance our promising therapeutic candidates,” said Ali Tehrani, Ph.D., President and Chief Executive Officer of Zymeworks. “These highly skilled individuals complement the team already in place as we work to bring new medicines to patients. With an expanding portfolio of clinical trials, increasing interaction with regulatory agencies, and scaling-up production of clinical materials, they will be able to make immediate and meaningful contributions to our accelerated development efforts.”
Senior Vice President, Technical and Manufacturing Operations
As Zymeworks’ Senior Vice President, Technical and Manufacturing Operations, Mr. Hollywood will be responsible for the manufacturing, quality, and regulatory compliance of Zymeworks’ lead assets as they advance towards potential commercialization. Mr. Hollywood brings 25 years of experience in the biopharmaceutical industry, most recently as Vice President and Head of ZymoGenetics, Inc. (a Bristol-Myers Squibb company) where he had overall management responsibility and led the business in supporting biologics portfolio deliverables including technical and manufacturing operations for development and commercial-stage programs.
Vice President, Clinical Research
As Vice President, Clinical Research, Dr. Josephson will lead the advancement of Zymeworks’ clinical pipeline as the Company prepares for the expansion of ZW25 into Europe and into the Asia-Pacific region with our recent partner BeiGene, Ltd., as well as the acceleration of clinical activities for ZW49. Before joining Zymeworks, Dr. Josephson was a Vice President of Clinical Development at Seattle Genetics, Inc. where he worked on multiple early and late-stage programs, including leading the front-line approval of ADCETRIS® for the treatment of Hodgkin’s lymphoma.
Vice President, Regulatory Affairs
As Vice President, Regulatory Affairs, Dr. Hart will be responsible for leading Zymeworks’ regulatory activities as the Company progresses into later stages of clinical development and expands development opportunities worldwide. Dr. Hart brings over 20 years of regulatory affairs experience, including 15 years at Seattle Genetics where he led the buildout of the regulatory affairs team and served as the regulatory lead for ADCETRIS® from the initiation of clinical testing through FDA approval.
Vice President, Business Development and Alliance Management
As Vice President, Business Development and Alliance Management, Dr. Poon will continue to be responsible for expanding Zymeworks’ strategic partnerships for therapeutic programs and platform technologies. Dr. Poon has held positions of increasing responsibilities during his 11-year tenure at Zymeworks and has been instrumental in establishing its strategic pharma and academic partnerships and collaborations, as well as overseeing competitive intelligence and alliance management.
Zymeworks is a clinical-stage biopharmaceutical company dedicated to the development of next-generation multifunctional biotherapeutics. The Company’s suite of therapeutic platforms and its fully integrated drug development engine enable precise engineering of highly differentiated product candidates. Zymeworks’ lead clinical candidate, ZW25, is a novel Azymetric™ bispecific antibody currently in Phase 2 clinical development. The Company’s second clinical candidate, ZW49, is a bispecific antibody-drug conjugate currently in Phase 1 clinical development and combines the unique design and antibody framework of ZW25 with Zymeworks’ proprietary ZymeLink™ cytotoxic payload. Zymeworks is also advancing a deep preclinical pipeline in immuno-oncology and other therapeutic areas. In addition, its therapeutic platforms are being leveraged through multiple strategic partnerships with eight global biopharmaceutical companies.
This press release includes “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. Forward-looking statements in this news release include, but are not limited to, statements that relate to the advancement of Zymeworks’ therapeutic candidates and assets through clinical development to commercialization, Zymeworks’ ability to bring new medicines to patients, the geographic expansion of Zymeworks’ activities, the potential for worldwide development opportunities, future contributions by the new Vice Presidents and other information that is not historical information. When used herein, words and phrases such as “advance”, “will,” “work to,” “progresses into” and similar expressions are intended to identify forward-looking statements. In addition, any statements or information that refer to expectations, beliefs, plans, projections, objectives, performance or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking. All forward-looking statements are based upon Zymeworks’ current expectations and various assumptions. Zymeworks believes there is a reasonable basis for its expectations and beliefs, but they are inherently uncertain. Zymeworks may not realize its expectations, and its beliefs may not prove correct. Actual results could differ materially from those described or implied by such forward-looking statements as a result of various factors, including, without limitation, market conditions and the factors described under “Risk Factors” in Zymeworks’ Annual Report on Form 10-K for its year ended December 31, 2018 (a copy of which may be obtained at www.sec.gov and www.sedar.com). Consequently, forward-looking statements should be regarded solely as Zymeworks’ current plans, estimates, and beliefs. Investors should not place undue reliance on forward-looking statements. Zymeworks cannot guarantee future results, events, levels of activity, performance or achievements. Zymeworks does not undertake and specifically declines any obligation to update, republish, or revise any forward-looking statements to reflect new information, future events or circumstances or to reflect the occurrences of unanticipated events, except as may be required by law.
Research Triangle Park, N.C., April 29, 2019 (GLOBE NEWSWIRE) — G1 Therapeutics, Inc. (Nasdaq: GTHX), a clinical-stage oncology company, today provided a regulatory update on trilaciclib, a first-in-class myelopreservation agent designed to protect the bone marrow from damage by chemotherapy and improve patient outcomes.
Based on
written feedback from its end-of-Phase 2 meeting with the U.S. Food and Drug
Administration (FDA) and discussions with European regulatory authorities, the
company plans to submit marketing applications in the U.S. and Europe for
trilaciclib for myelopreservation in small cell lung cancer (SCLC). These
submissions will be based on currently available data from three randomized,
double-blind, placebo-controlled SCLC clinical trials, as well as safety data
collected across all completed and ongoing clinical trials.
“We are
pleased with the feedback from our recent meetings with regulatory authorities.
We look forward to continuing a collaborative dialogue regarding the marketing
applications, as well as discussing further clinical development of
trilaciclib,” said Raj Malik, M.D., Chief Medical Officer. “We believe
trilaciclib represents an important advance in the care of SCLC patients. We
will also move forward with a robust development program to evaluate
trilaciclib in multiple tumor types and chemotherapy regimens.”
G1 will
request a pre-New Drug Application (NDA) meeting with the FDA and anticipates
it will be scheduled later this year. The company will provide further details
regarding the NDA submission and timeline following that meeting. The company
plans to submit a Marketing Authorization Application (MAA) to the European
Medicines Agency (EMA) subsequent to an NDA filing.
Chemotherapy
is an effective and important weapon against cancer. However, chemotherapy does
not differentiate between healthy cells and cancer cells and kills both,
including important stem cells in the bone marrow that produce white blood
cells, red blood cells and platelets. This chemotherapy-induced bone marrow
damage is known as myelosuppression. When white blood cells, red blood cells
and platelets become depleted, chemotherapy patients are at increased risk
of infection, experience anemia and fatigue, and are at increased risk of
bleeding. Myelosuppression often requires the administration of rescue
interventions such as growth factors and blood or platelet transfusions, and
may also result in chemotherapy dose delays and reductions.
“In clinical
trials, trilaciclib demonstrated the ability to protect bone marrow from
chemotherapy damage and meaningfully reduced the need for supportive care
interventions, such as G-CSF and transfusions,” said Jared Weiss, M.D.,
Associate Professor, University of North Carolina Lineberger Comprehensive
Cancer Center, and trilaciclib clinical trial investigator. “By providing
a proactive approach to reduce myelosuppression, trilaciclib improves the
patient experience during chemotherapy treatment, reducing side effects and the
need for associated interventions commonly given to treat them.”
About Trilaciclib Trilaciclib is a first-in-class myelopreservation agent designed to protect the bone marrow from damage by chemotherapy and improve patient outcomes. Trilaciclib is being evaluated in four randomized Phase 2 clinical trials; G1 reported positive results from all these trials in 2018.
About G1 Therapeutics G1 Therapeutics, Inc. is a clinical-stage biopharmaceutical company focused on the discovery, development and delivery of innovative therapies that improve the lives of those affected by cancer. The company is advancing three clinical-stage programs. Trilaciclib and lerociclib are designed to enable more effective combination treatment strategies and improve patient outcomes across multiple oncology indications. G1T48 is a potential best-in-class oral selective estrogen receptor degrader (SERD) for the treatment of ER+ breast cancer. G1 also has an active discovery program focused on cyclin-dependent kinase targets.
Forward-Looking
Statements
This press release contains forward-looking statements within the meaning of
the Private Securities Litigation Reform Act of 1995. Words such as
“may,” “will,” “expect,” “plan,”
“anticipate,” “estimate,” “intend” and similar
expressions (as well as other words or expressions referencing future events,
conditions or circumstances) are intended to identify forward-looking
statements. Forward-looking statements in this news release include, but are
not limited to, the therapeutic potential of trilaciclib, lerociclib and G1T48
and the timing for next steps with regard to the trilaciclib marketing applications,
and are based on the Company’s expectations and assumptions as of the date of
this press release. Each of these forward-looking statements involves risks and
uncertainties. Factors that may cause the Company’s actual results to differ
from those expressed or implied in the forward-looking statements in this press
release are discussed in the Company’s filings with the U.S. Securities and
Exchange Commission, including the “Risk Factors” sections contained
therein and include, but are not limited to, the Company’s ability to complete
clinical trials for, obtain approvals for and commercialize any of its product
candidates; the Company’s initial success in ongoing clinical trials may not be
indicative of results obtained when these trials are completed or in later
stage trials; the inherent uncertainties associated with developing new
products or technologies and operating as a development-stage company; the
Company’s development of a CDK4/6 inhibitor to reduce chemotherapy-induced
myelosuppression is novel, unproven and rapidly evolving and may never lead to
a marketable product; and market conditions. Except as required by law, the
Company assumes no obligation to update any forward-looking statements
contained herein to reflect any change in expectations, even as new information
becomes available.
Expansion of ongoing AVID200 Phase 1 solid tumor trial to include patients at clinical sites in Canada
AVID200 is a rationally designed, highly potent inhibitor of TGF-beta 1 & 3, the principal oncogenic TGF-beta isoforms
Best-in-class efficacy and safety potential by selectively targeting TGF-beta 1 & 3 while sparing TGF-beta 2, the isoform that promotes normal cardiac function
Austin, TX, and Montreal, QC (Apr. 29, 2019) – Forbius, a clinical-stage company that develops novel biologics for the treatment of cancer and fibrosis, announced that it has received a no objection letter from Health Canada for its clinical trial application (CTA) to conduct a Phase 1 trial in solid tumors with immuno-oncology candidate AVID200, a rationally designed and highly potent inhibitor of TGF-beta 1 & 3.
AVID200-03 (NCT03834662) is a Phase I, open-label, dose-escalation trial to establish the recommended phase 2 dose (RP2D) of AVID200 in patients with advanced or metastatic malignancies. The trial is currently enrolling patients at centers in the U.S. and will now be expanded to additional clinical sites in Canada to recruit a total of up to 36 patients.
TGF-beta 1 & 3 are the main oncogenic TGF-beta isoforms expressed by many solid tumors and represent promising immuno-oncology targets. These TGF-beta isoforms are implicated in T-cell suppression, fibrosis in the tumor microenvironment, and resistance to immunotherapeutics such as nivolumab (Opdivo) and pembrolizumab (Keytruda) (Chakravarthy et al., Nature Comm., 2018; Tauriello et al., Nature, 2018; Mariathasan et al., Nature, 2018).
About AVID200 and the AVID200-03 Trial
AVID200 is an isoform-selective and highly potent inhibitor of TGF-beta 1 & 3 undergoing Phase 1 clinical testing in solid tumors and fibrotic diseases. TGF-beta 1 & 3 are the principal disease-driving isoforms, while TGF-beta 2 is responsible for normal cardiac function and hematopoiesis.
AVID200’s selectivity for TGF-beta 1 & 3 was designed to achieve optimal efficacy while circumventing cardiac and other safety issues that have limited the applicability of older-generation, non-selective TGF-beta inhibitors. Therefore, AVID200 is positioned to be an effective and well-tolerated therapeutic in a variety of clinical settings, including in combination with anti-PD-(L)1 therapy.
AVID200-03 (NCT03834662) is an open label, multicenter, dose-escalation study to evaluate the safety, pharmacokinetics, pharmacodynamics, and antitumor effects of AVID200 in patients with advanced or metastatic solid tumor malignancies.
About Forbius
Forbius is a clinical-stage protein engineering company that designs and develops novel biologics for the treatment of fibrosis and cancer. Our current focus is the development of agents targeting the transforming growth factor-beta (TGF-beta) and epidermal growth factor receptor (EGFR) pathways.
MANNHEIM, Germany – April 26, 2019 — Cardiac Dimensions, a leader in the development of innovative, minimally invasive treatments to address functional mitral regurgitation (FMR) in patients with heart failure (HF), presented new data confirming significant reduction in regurgitant volume directly after implantation of the Carillon® Mitral Contour System® at the German Society of Cardiology annual congress, DGK 2019. The Carillon® System is a right-heart transcatheter mitral valve repair (TMVr) device designed to treat the primary cause of FMR in patients with MR grades 2+, 3+ and 4+.
Prof. Dr. Andreas Hagendorff, Head of Laboratory of Echocardiography of the Department of Cardiology-Angiology at the University of Leipzig and renowned echo-cardiologist led the study. “Functional mitral regurgitation is associated with increased morbidity and mortality in patients with heart failure. For patients ineligible to receive surgical treatment, interventional techniques represent a new therapy option.” Prof. Hagendorff continued, “The findings we presented confirmed the acute hemodynamic effects of the Carillon® device on mitral valve morphology and demonstrated an acute reduction in a regurgitant fraction in 83% of patients, as documented by transthoracic echo after device implantation.”
“The significant acute MR benefit demonstrated in our study, achieved by a device that cinches the mitral apparatus via the coronary sinus without compromising the valve or future treatment options, makes the Carillon® System a front-line treatment option for FMR patients,” added Dr. Stephan Stoebe, co-author and presenter at DGK.
Presentations highlighting the Carillon® System this week during DGK 2019 include:
25 April 2019: New Devices and Therapies for Mitral Valve Insufficiency Treatment of functional mitral regurgitation by the Carillon® Mitral Contour Device – an echocardiographic analysis of acute effects. S. Stöbe, K. Kreyer, U. Laufs, A. Hagendorff
26 April 2019: Functional MI in Heart Failure: Interventional Differential Therapie Background on the Carillon® Mitral Contour System® & REDUCE FMR. H. Sievert Carillon® Mitral Contour System® Live Case. M. Haude Atrium-Triggered Annulus-Extension. M. Reinthaler Combo-Cases – MitraClip & Carillon, Carillon® & MitraClip R. S. von Bardeleben (select presentations from DGK 2019 can be found at DGK 2019 Presentations)
“We are pleased with the results from the University of Leipzig study validating the immediate impact of the Carillon® System on mitral regurgitation for a large percentage of FMR patients,” said Greg Casciaro, President and CEO of Cardiac Dimensions. “This study adds to the growing body of evidence demonstrating the effectiveness and safety of the Carillon System in two key measurements of FMR – a significant MR reduction and favorable left ventricular remodeling, observed most recently in the REDUCE FMR trial, the first and only randomized, blinded, sham-controlled trial conducted in structural heart.”
About the Carillon® Mitral Contour System The Carillon® System offers a simple right-heart approach to transcatheter mitral valve repair (TMVr) designed to reshape the anatomy and improve the function of the mitral apparatus from the coronary sinus. Distal and proximal anchors, connected by a shaping ribbon, utilize the heart’s venous anatomy to cinch the mitral apparatus, without compromising the valve or future treatment options.1,2 The Carillon® System is designed to treat the primary cause of functional mitral regurgitation (FMR) in patients with MR grades 2+, 3+ and 4+ and is the first and only device to demonstrate a reduction in regurgitant volume and reverse left ventricular remodeling in a randomized sham-controlled clinical trial of percutaneous valve therapy 3,4,5.
The Carillon® System has CE Mark (0344) approval and has been implanted in over 950 patients in Europe, Australia, Turkey, and the Middle East. The Carillon® System is not approved for sale in the United States.
About Cardiac Dimensions, Inc. Cardiac Dimensions is a leader in the development of innovative, minimally invasive treatments to address heart failure and related cardiovascular conditions. Privately held, the company is funded by Aperture Venture Partners, Arboretum Ventures, Difference Capital, HostPlus, Life Sciences Partners, Lumira Ventures and M.H. Carnegie & Co. Cardiac Dimensions is headquartered in Kirkland, Washington and has operations in the United States, Australia, and Germany.
1. Hoppe UC, Brandt MC, Degen H, et al. Percutaneous mitral annuloplasty device leaves free access to cardiac veins for resynchronization therapy. Catheter Cardiovasc Interv. 2009;74(3):506-11. 2. Latib, A. “Coronary Sinus Annuloplasty.” New York, Montefiore Medical Center. 3. Lipiecki J, Siminiak T, Sievert H, et al. Coronary sinus-based percutaneous annuloplasty as treatment for functional mitral regurgitation: the TITAN II trial. BMJ Open Heart. 2016; 3: e000411. 4. Siminiak T, et. al. Treatment of functional mitral regurgitation by percutaneous annuloplasty: Results of the TITAN Trial. Eur J Heart Fail. 2012;14:931-38. 5. Sievert, H. 2018. REDUCE-FMR: A Sham Controlled Randomized Trial of Transcatheter Indirect Mitral Annuloplasty in Heart Failure Patients with Functional Mitral Regurgitation. Presented at TCT 2018, San Diego, CA.
Funding supports Phase 3 development of STS101 for the acute treatment of migraine, including pivotal Phase 3 efficacy trial with planned initiation in Q3 2019
SOUTH SAN FRANCISCO, Calif., April 24, 2019 /PRNewswire/ — Satsuma Pharmaceuticals, Inc. (“Satsuma” or “the Company”), a clinical-stage biopharmaceutical company developing STS101, (dihydroergotamine (DHE) nasal powder) for the acute treatment of migraine, today announced a $62 million Series B preferred stock financing led by Wellington Management Company, with participation by existing investors RA Capital Management, TPG Biotech, and Shin Nippon Biomedical Laboratories, and new investors, Osage University Partners, CAM Capital, Surveyor, Eventide Asset Management, Cormorant, Lumira Ventures, and SBI Investment.
Financing proceeds will support Phase 3 development of STS101. In consultation with the FDA, Satsuma has established key elements of its Phase 3 development program, including the design of the Phase 3 efficacy study, to support a New Drug Application (NDA) for STS101 for the acute treatment of migraine. Satsuma plans to initiate its randomized, double-blind, placebo-controlled Phase 3 clinical trial of STS101 in the third quarter of 2019.
John Kollins, President and Chief Executive Officer of Satsuma, commented, “As we advance STS101 into Phase 3 development, we are privileged to have strong support from top-tier healthcare investors who share our vision of creating a best-in-class DHE therapeutic product with differentiated and demonstrated clinical benefits that address the unmet needs of many people with migraine.”
About Satsuma Pharmaceuticals
Satsuma Pharmaceuticals is a clinical-stage biopharmaceutical company focused on developing STS101 as an important and differentiated therapeutic option for the acute treatment of migraine. STS101 is a novel and proprietary investigational drug-device combination product specifically designed to enable intranasal administration of the anti-migraine drug, dihydroergotamine (DHE), with a pharmacokinetic profile optimized to provide consistent and robust clinical efficacy. In developing STS101, Satsuma has applied proprietary nasal drug delivery, dry-powder formulation, and engineered drug particle technologies to create a compact, simple-to-use, self-administered, and non-injectable DHE product. The Company believes STS101 will be an attractive migraine treatment option for many patients and may enable a larger number of people with migraine to realize the long-recognized therapeutic benefits of DHE therapy. STS101 has undergone extensive pre-clinical optimization and recently completed a Phase 1 clinical trial.
Satsuma is headquartered in South San Francisco, California with operations in both California and Research Triangle Park, North Carolina.
CORPORATE CONTACTS:
Tom O’Neil, Chief Financial Officer Satsuma Pharmaceuticals, Inc. tom@satsumarx.com
John Kollins, President and Chief Executive Officer Satsuma Pharmaceuticals, Inc. john@satsumarx.com
LISLE, Ill., April 24, 2019 /PRNewswire/ — Endotronix, Inc., a digital health and medical technology company dedicated to advancing the treatment of heart failure, today announced that the company received ISO 13485:2016 certification from British Standards Institution (BSI) for its Quality Management System (QMS). Endotronix’s adherence to internationally recognized standards for the medical device industry demonstrates its commitment to the highest level of product quality. They have developed a comprehensive heart failure solution, the Cordella™ Heart Failure System (Cordella System), which consists of a remote patient management platform and the implantable Cordella™ Pulmonary Artery Pressure Sensor (Cordella Sensor).
“We are focused on executing several key business, regulatory, and clinical milestones this year that ultimately drive the success of the company. The recent ISO certification ensures we are fully aligned with global regulatory requirements to provide a best-in-class product quality experience to our customers,” commented Harry Rowland, CEO of Endotronix. “Similarly, we continue to make excellent progress with our comprehensive clinical program, which will show the real-world benefits of the Cordella System and create U.S. and E.U. market access for the Cordella Sensor.”
The Cordella System is a comprehensive heart failure management solution that enables proactive management and early detection of worsening heart failure. The system can be seamlessly integrated with a next generation pulmonary artery (PA) pressure sensor, the Cordella Sensor, to streamline heart failure care management, reduce heart-failure-related hospitalizations, and support reimbursement for care delivery activities.
The Cordella System is available for commercial use in the U.S. and E.U. and is currently in cardiology centers across the U.S. The Cordella Sensor is an investigational device and is not available for commercial use in any geography.
About Endotronix
Endotronix, Inc., a medical technology company, delivers an integrated platform that provides comprehensive, reimbursable health management innovations for patients suffering from chronic heart failure. Their solution, the Cordella™ Heart Failure System, includes a cloud-based disease management data platform and at-home hemodynamic management with a breakthrough implantable wireless pulmonary artery pressure sensor for early detection of worsening heart failure.
MEDIA CONTACT: Carla Benigni SPRIG Consulting, LLC +1 (847) 951-7430
Investigator-initiated trial to evaluate AVID200’s safety, anti-fibrotic activity, and ability to restore hematopoiesis in patients with myelofibrosis
AVID200 is a rationally designed, highly potent inhibitor of TGF-beta 1 & 3, the principal drivers of fibrosis in myelofibrosis and other fibrotic diseases
AVID200 spares TGF-beta 2, the isoform that promotes hematopoiesis and normal cardiac function
Austin, TX and Montreal, QC (Apr. 24, 2019) – Forbius, a clinical-stage company that develops novel biologics for the treatment of fibrosis and cancer, announced today that the first patient has been dosed in a Phase 1b trial assessing AVID200, a novel TGF-beta 1 & 3 inhibitor, in patients with myelofibrosis (MF). This multicenter trial is sponsored by the Icahn School of Medicine at Mount Sinai and the Myeloproliferative Neoplasm Research Consortium (MPN-RC), with the support of a peer-reviewed NIH grant.
AVID200’s novel dual mode of action in MF centers around its anti-fibrotic effects and ability to restore hematopoiesis, as demonstrated in MF patient cells and in vivo models of the disease (Varricchio et al., 2018). Notably, treatment of cells from MF patients with AVID200 promoted proliferation of normal hematopoietic progenitors while decreasing the proportion of MF malignant progenitor cells.
“This clinical trial will evaluate the ability of AVID200 to achieve the disease-modifying outcomes of reversing bone marrow fibrosis and restoring normal hematopoiesis. Preclinical data demonstrate that selective neutralization of TGF-beta 1 & 3 by AVID200 results in both of these critical outcomes. We believe that AVID200 has the potential to become the first disease-modifying treatment for MF,” commented Dr. Ronald Hoffman, founder of the MPN-RC and Director of the Myeloproliferative Disorders Research Program at the Icahn School of Medicine at Mount Sinai.
The single-arm, dose-escalation Phase 1b trial (AVID200-02; NCT03895112) plans to enroll up to 24 patients with primary MF, post-essential thrombocythemia MF, or post-polycythemia vera MF with > grade 2 bone marrow fibrosis. Outcome measures include safety, response, clinical and hematological improvement, as well as assessment of the degree of bone marrow fibrosis.
About AVID200 and the AVID200-02 Trial
AVID200 is an isoform-selective and highly potent inhibitor of TGF-beta 1 & 3, the two principal pro-fibrotic TGF-beta isoforms. These TGF-beta isoforms are central regulators in the pathogenesis and progression of fibrotic diseases, including MF (Chagraoui et al., 2002). AVID200 was rationally designed to be minimally active against TGF-beta 2, which is a promoter of hematopoiesis and normal cardiac function. This optimal selectivity positions AVID200 to be an effective and well-tolerated therapeutic for MF and other fibrotic diseases.
AVID200-02 (NCT03895112) is an investigator-initiated, open-label, multicenter, Phase 1b trial to evaluate the safety and anti-fibrotic activity of AVID200, as well as its ability to restore normal hematopoiesis in patients with MF.
About the Myleoproliferative Neoplasm Research Consortium (MPN-RC)
The MPN-RC was founded in 2006 and is the only independent, multicenter, international consortium of scientists and clinicians dedicated to developing novel therapeutic strategies for MF and other myeloproliferative neoplasms (MPN). The MPN-RC is funded by the NIH to conduct clinical trials based on the most promising preclinical MPN research. The goal of the consortium is to adapt quickly in response to scientific advances and a changing clinical landscape, in order to develop effective therapeutics for MPN patients.
About Myelofibrosis (MF)
MF is a rare, life-threatening blood cancer characterized by progressive bone marrow fibrosis, which causes ineffective hematopoiesis. Approximately 30,000 people in the US alone are affected by this disease. Currently, there are no approved therapies targeting the underlying bone marrow fibrosis available to MF patients.
About Forbius
Forbius is a clinical-stage protein engineering company that designs and develops novel biologics for the treatment of fibrosis and cancer. Our current focus is the development of agents targeting the transforming growth factor-beta (TGF-beta) and epidermal growth factor receptor (EGFR) pathways.
A First-in-Class, Non-Surgical Solution for Obesity Approved for One-Year of Device Treatment, Offering Patients Potentially More Durable Weight Loss and Health Benefits
SAN CARLOS, Calif., April 23, 2019 /PRNewswire/ — BAROnova, Inc., a medical device company focused on the development of first-in-class non-surgical solutions for the treatment of obesity, announced today that the U.S. Food and Drug Administration (FDA) has approved the TransPyloric Shuttle® (TPS®) Device , a non-surgical weight loss solution for adult individuals suffering from obesity with a body mass index (BMI) of 30 to 40 kg/m2. The device is approved for up to 12 months of treatment, during which time patients undergo lifestyle modification counseling to help develop and maintain healthier habits.
The TPS® device is delivered and retrieved endoscopically and relies on its mechanical structure to maintain its shape to keep it in the stomach as opposed to traditional inflatable intragastric balloons. The constructed TPS® is the size of a small peach (~5.6 cm in diameter), 85-90% smaller than the fluid-filled balloons. Once in stomach, it is designed to slow the passage of food so patients feel full sooner and stay full longer. The 12-month treatment duration is twice as long as intragastric balloons currently approved in the U.S. The longer treatment duration potentially allows patients the opportunity to achieve more durable lifestyle changes and health benefits.
“Those of us who participated in the pivotal trial have the firsthand experience with the TPS® device and were pleased with its patient outcomes. In addition to the clinically meaningful weight loss seen in the study group who received the device, we also observed cardio-metabolic improvements in that cohort. We are excited to see that the device is approved by the FDA and look forward to using it in our clinical practice.” said Dr. Richard Rothstein, the Joseph M. Huber Professor and Chair of Medicine for the Dartmouth Geisel School of Medicine, and the lead investigator for the ENDObesity II study.
Obesity is one of the
biggest drivers of the healthcare costs in the U.S., currently estimated
as much as $210 billion annually. Effectively treating this condition will have
a substantial impact on controlling ever-increasing healthcare costs.
“The vast majority of
patients with obesity are left untreated today. Endoscopically delivered
intragastric devices can help close the obesity treatment gap and offer
alternative options for qualified patients who are not eligible, or unwilling,
to undergo metabolic and bariatric surgery. The TPS device design addresses
some of the limitations with the first-generation intragastric devices and
offers longer treatment duration which is clinically attractive.” commented Dr.
Wayne English, Associate Professor of Surgery, Clinical Trial Research Director
at Vanderbilt University Weight Loss Center, and a principal investigator for
the ENDObesity II study.
“FDA approval is a significant milestone for the company.” said Lian Cunningham, M.D, PhD, Senior Vice President of Clinical Affairs and Regulatory Affairs of BAROnova Inc. “The TPS® device is unique in its treatment efficacy and duration. Further validation of potential comorbidity benefits, like cardiometabolic improvement observed in the pivotal study, during the post-market phase may translate to important advantages in achieving healthcare coverage for the TPS® device in the future. We look forward to bringing this new technology to patients and physicians.”
The approval of the TPS® is based on data from its pivotal trial (ENDObesity® II study), a randomized, double-blind, and sham-controlled study that enrolled 302 patients from nine investigational centers across the United States. Patients treated with the TPS® device on average lost 3.4x more weight when compared to the sham-control group (9.5% for the TPS® group and 2.8% for the Control, p<0.0001) at the 12-month follow up. Approximately 67% of people treated with TPS® lost 5% or more of their body weight, exceeding the study endpoint target of 50% (p<0.0001). Forty percent (40%) of people treated with TPS® lost 10% or more weight (vs. 14% in sham-treated controls). A weight loss of 5% or more is considered clinically meaningful for achieving important health benefits. Improvements in blood pressure and other cardiometabolic risk factors as well as quality of life were also observed with TPS® treatment. The most common adverse events among people treated with the TPS® device were gastrointestinal events, such as stomach pain, nausea, vomiting, and dyspepsia, as expected with an intragastric device designed to treat obesity through delayed gastric emptying.
About Obesity
Obesity
is defined as a Body Mass Index (BMI) >30 kg/m2. Individuals with
obesity are at increased risk of developing over 70 comorbid conditions
including hypertension, hyperlipidemia, and type 2 diabetes. Obesity is a
worldwide epidemic that has placed a major burden on healthcare systems
globally. There are currently over 75 million adults in the United States
with a BMI between 30-40 kg/m2 and over 600 million adults worldwide
who are candidates for a safe and effective non-surgical therapy.
About the TransPyloric Shuttle®(TPS®)
TPS® is a novel device that is designed to be inserted and removed trans-orally using standard endoscopic techniques. It is mechanically constructed using solid silicone components and is not subject to inflation or deflation risks. The TPS/TPS® Delivery Device is indicated for weight reduction in adult patients with obesity with a Body Mass Index (BMI) of 35.0-40.0 kg/m2 or a BMI of 30.0 to 34.9 kg/m2 with one or more obesity-related comorbid conditions and is intended to be used in conjunction with a diet and behavior modification program. The device is intended to reside in the stomach for 12 months. The TransPyloric Shuttle®’s primary mechanism of action is delayed gastric emptying which is a known mechanism of weight loss.
About BAROnova, Inc.
BAROnova is a private medical device company that has developed a first-in-class non-surgical solution for obesity. BAROnova is headquartered in San Carlos, CA.
This trial evaluates the efficacy of AVID100 in TNBC patients with EGFR-overexpression
20% of TNBC patients highly overexpress EGFR; there is no approved targeted therapy
AVID100 is the most advanced, broadly active anti-EGFR ADC in clinical development, targeting both wild-type and mutant forms of EGFR
Austin, TX, and Montreal, QC (Apr. 22, 2019) – Forbius, a clinical-stage company that develops novel biologics for the treatment of cancer and fibrosis, announced today that the first patient has been dosed in a Phase 2a triple negative breast cancer (TNBC) clinical trial with AVID100, a novel, tumor-selective anti-epidermal growth factor receptor (EGFR) antibody-drug conjugate (ADC).
Approximately 20% of TNBC patients have tumors that highly overexpress EGFR. No targeted therapy is approved for EGFR-overexpressing TNBC.
The multicenter, dose-expansion Phase 2a trial (AVID100-01; NCT03094169) will evaluate the efficacy, safety, and tolerability of AVID100 in patients with advanced, EGFR-overexpressing TNBC (IHC 2+/3+). This is the third cohort that has been launched and follows the previously announced cohorts evaluating AVID100 in patients with advanced squamous non-small cell lung cancer (sqNSCLC) and squamous cell carcinoma of the head and neck (SCCHN). In total, approximately 100 patients will be evaluated across three EGFR-overexpressing tumor types: sqNSCLC, SCCHN, and TNBC.
About AVID100 and the AVID100-01 Trial
AVID100 is a highly potent EGFR-targeting ADC engineered to achieve enhanced anti-tumor efficacy without a corresponding increase in toxicity in skin or other EGFR-expressing normal tissues. In preclinical studies, AVID100 demonstrated significant anti-cancer activity in EGFR-overexpressing tumor models resistant to marketed EGFR inhibitors. AVID100 is the most advanced, broadly active anti-EGFR ADC in clinical development and targets both wild-type and mutant forms of EGFR.
A recommended Phase 2 dose (RP2D) of 220 mg/m2 (~6mg/kg) was established for AVID100 in a completed Phase 1 study. This RP2D is expected to be in the therapeutically active range based on preclinical efficacy studies. The majority of treatment-related adverse events in the Phase 1 trial at the RP2D were well-tolerated and grade 1 or 2 in severity.
AVID100-01 (NCT03094169) is an open-label, multicenter, dose-expansion study to evaluate the efficacy, safety, and tolerability of AVID100 in patients with confirmed EGFR-overexpressing sqNSCLC (IHC 3+), SCCHN (IHC 3+), and TNBC (IHC 2+/3+) (more than 50% of cells with EGFR 3+ or more than 75% of cells with EGFR 2+ staining).
About Forbius
Forbius is a clinical-stage protein engineering company that designs and develops novel biologics for the treatment of fibrosis and cancer. Our current focus is on the development of agents that target the transforming growth factor-beta (TGF-beta) and the epidermal growth factor receptor (EGFR) pathways.
SEATTLE, April 16, 2019 /PRNewswire/ — Bardy Diagnostics, Inc., (“BardyDx”), a leading provider of ambulatory cardiac monitoring technologies and custom data solutions, announced today it has raised $35.5 million in Series B funding, led by River Cities Capital Funds with participation from new investors HealthQuest Capital, Aperture Venture Partners, Aphelion Capital, Lumira Ventures, and Rex Health Ventures. Existing equity investors, SV Health Investors, Health Enterprise Partners, and Ascension Ventures also joined the round. The funds will be used to further accelerate growth of its innovative P-wave focused cardiac monitoring platform through expansion of BardyDx’s sales force and monitoring services, and to support advanced development programs, including augmented intelligence and visualization technologies.
“The caliber of our new investors in addition to our exceptional existing syndicate of financiers underscores the exciting opportunity before us,” said Gust H. Bardy, MD, Founder and Chief Executive Officer of BardyDx. “With these funds, we will grow our business by refining and expanding our primary goal of revealing to patients, and their physicians, the full complexity and meaning of their cardiac rhythm.”
BardyDx’s Carnation Ambulatory Monitor (“CAM™”) is the industry’s only P-wave centric™ ambulatory cardiac patch monitor and arrhythmia detection device. The genesis of the CAM is a result of Dr. Bardy’s frustrations with the fundamental flaws rooted in the development and evolution of cardiac monitoring.
“For nearly 60 years, standard ECG engineering practices have over-processed the heart’s electrical message to make machines’ ‘lives’ easier at the expense of losing details in the ECG,” added Dr. Bardy. “In essence, such legacy engineering doesn’t listen to what the heart is trying to tell us but rather tells the heart what we are willing to hear. At BardyDx, our pioneering technology focuses on revealing the cardiac rhythm message fully, and in so doing, allows for precise and meaningful patient management predicated on cardiac truth.”
BardyDx also announced that Rik Vandevenne, Managing Director of River Cities Capital Funds, and Garheng Kong, Managing Director of HealthQuest Capital, will join the company’s board of directors.
“It’s not every day you have the opportunity to partner with such a groundbreaking serial entrepreneur and innovator as Dr. Gust Bardy and the team at BardyDx,” said Vandevenne. “The company has a clear focus on putting the patient first and they have developed a clinically-superior ECG platform that is revolutionizing heart monitoring. We are excited for the opportunity to help further drive growth for the company while positively impacting the lives of thousands of people every week.”
About Bardy Diagnostics:
Bardy Diagnostics, Inc. is an innovator in digital health and remote patient monitoring, with a focus on providing the most diagnostically-accurate and patient-friendly cardiac patch and other monitors to the industry. The company’s CAM patch is a non-invasive, P-wave centric™ ambulatory cardiac monitor and arrhythmia detection device that is designed to improve patient compliance for adults and children weighing more than 10kg (22lbs) through its lifestyle-enabling design. Designed to be worn comfortably and discreetly, the female-friendly, hourglass-shaped CAM patch is placed on the center of the chest, directly over the heart for optimum ECG signal collection. The proprietary technology of the CAM patch provides optimal detection and clear recording of the often difficult-to-detect P-wave, the signal of the ECG waveform that is essential for accurate arrhythmia diagnosis. For more information, please visit www.bardydx.com.
About River Cities Capital Funds:
River Cities is a growth equity firm investing in high-potential healthcare and information technology companies. A consistent, cohesive team has honed its strategy over six funds with compelling performance. River Cities seeks to be a business partner first and a capital provider second, investing significant human capital to leverage its domain expertise and a network of thought leaders assembled over the last 25 years. With $750 million of capital raised and a consistent track record of success, River Cities has established itself as a preferred source of growth capital. The firm, located in Cincinnati, OH and Raleigh, NC, is actively seeking new investments for its Fund VI. For more information, please visit www.rccf.com.
MEDIA CONTACT:
Jonathan Wu Director, Marketing Bardy Diagnostics, Inc. 1-844-422-7393 jwu@bardydx.com
VICTORIA, British Columbia–(BUSINESS WIRE) —Aurinia Pharmaceuticals Inc. (NASDAQ: AUPH / TSX: AUP) (“Aurinia” or the “Company”), a late clinical-stage biopharmaceutical company focused on the global immunology market, today announced the appointment of Mr. Peter Greenleaf as Chief Executive Officer and as a Director on the Aurinia Board. The Company also announced the elevation of George M. Milne, Jr., Ph.D. to Chairman of the Board of Directors. Dr. Richard M. Glickman, who previously announced his plans to retire on November 6, 2018, will step down from his role as Chairman and CEO concurrent with Mr. Greenleaf’s appointment on April 29, 2019, and will remain an advisor to the Company for a period of 12 months.
With more than twenty years of experience leading pharmaceutical and biotech firms, Mr. Greenleaf most recently served as the CEO of Cerecor, a leading U.S. pediatric orphan and rare disease pharmaceutical company. Prior to that, Mr. Greenleaf was the Chairman and CEO of Sucampo Pharmaceuticals which he led through the successful sale to Mallinckrodt Pharmaceuticals, PLC for $1.2B. Previously, Mr. Greenleaf served as the CEO and Board member of Histogenics, a regenerative medicine company. Prior to that he was the President of MedImmune, Inc, the global biologics arm of AstraZeneca, and President of MedImmune Ventures, a wholly owned venture capital fund within the AstraZeneca Group, where he led investment in emerging biopharmaceutical, medical device, and diagnostic companies.
“It is a pleasure to welcome Peter as the next Chief Executive Officer of Aurinia. As a seasoned leader in the pharmaceutical industry, Peter’s extensive knowledge of clinical and overall operations, along with business development and commercialization expertise, are ideally aligned with the next stages of growth for voclosporin and Aurinia,” stated Dr. George Milne, incoming Chairman of the Board of Aurinia Pharmaceuticals.
“The Aurinia team has made extraordinary progress with voclosporin, which I believe to be a truly transformative drug for the potential treatment of proteinuric kidney diseases, such as lupus nephritis (“LN”), as well as a unique opportunity for the treatment of dry eye syndrome (“DES”),” commented Mr. Greenleaf. “To that end, I am very excited to lead the Company at this pivotal time and through several critical datapoints over the next year including Phase 3 trial results by the end of 2019, followed by the planned regulatory submission and preparations for the potential commercialization of voclosporin during 2020.”
Dr. Milne further commented, “I am also humbled to be assuming the Chairman role from Dr. Glickman. On behalf of the entire board and organization, I would like to thank Dr. Glickman for all of his efforts and contributions that have brought Aurinia to where it is today. As we wish him all the best on his retirement, we are also gratified to have his insight as an advisor for the next year.”
Dr. Glickman stated, “Consistent with the succession planning set into motion last November, I am confident that Peter is the correct individual to lead Aurinia through the next set of value inflection points including the upcoming AURORA Phase 3 results, preparing for the potential launch of voclosporin, and expansion of the VOS dry eye syndrome program.”
About Mr. Peter Greenleaf
Peter Greenleaf previously served as CEO of Cerecor, Inc., since March 2018. Prior to that he served as Chairman and CEO of Sucampo Pharmaceuticals, Inc. from March 2014 to February 2018, when Sucampo was sold to Mallinckrodt PLC. Previously, Mr. Greenleaf served as CEO of Histogenics Corporation from June 2013 to March 2014, as President of MedImmune, Inc., and MedImmune Ventures from 2010 to June 2013, and Senior Vice President, Commercial Operations of MedImmune from 2006 to 2010. Mr. Greenleaf also held senior commercial roles at Centocor Biotech, Inc. (now Janssen Biotechnology, Johnson & Johnson), from 1998 to 2006, and at Boehringer Mannheim G.m.b.H. (now Roche Holdings) from 1996 to 1998. Mr. Greenleaf is a member of the Board of Directors of Cerecor since May 2017, is the Chairman of the Board of Bio-delivery Sciences since May 2018, EyeGate Pharmaceuticals since August 2018, and Antares Pharma since December 2018. Mr. Greenleaf chairs the Maryland Venture Fund Authority, and previously served on the boards of BIO, PhARMA, the Tech Council of Maryland and the University of Maryland Baltimore Foundation, Inc. Mr. Greenleaf earned an MBA degree from St. Joseph’s University and a BS degree from Western Connecticut State University.
About George M. Milne, Jr., Ph.D.
Dr. Milne was appointed to the Aurinia Board of Directors in May 2017 and serves as Chair of the Company’s Governance & Nomination Committee. Dr. Milne has over 30 years of experience in pharmaceutical research and product development. He joined Pfizer in 1970 and held a variety of positions conducting both chemistry and pharmacology research. Dr. Milne became director of the department of immunology and infectious diseases at Pfizer in 1981, was its executive director from 1984 to 1985, and was vice president of research and development from 1985 to 1988. He was appointed senior vice president in 1988. In 1993 he was appointed President of Pfizer Central Research and a senior vice president of Pfizer with global responsibility for human and veterinary medicine research and development. Dr. Milne has served on multiple corporate boards including Mettler-Toledo, Inc., MedImmune, Athersys, Biostorage Technologies, Aspreva, and Conor Medsystems. Dr. Milne received his B.Sc. in Chemistry from Yale University and his Ph.D. in Organic Chemistry from MIT.
About Aurinia
Aurinia Pharmaceuticals is a late clinical-stage biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are impacted by serious diseases with a high unmet medical need. The Company is currently developing voclosporin, an investigational drug, for the potential treatment of LN, Focal Segmental Glomerulosclerosis (“FSGS”), and DES. The Company is headquartered in Victoria, British Columbia and focuses its development efforts globally.
About Voclosporin
Voclosporin, an investigational drug, is a novel and potentially best-in-class calcineurin inhibitor (“CNI”) with clinical data in over 2,400 patients across indications. Voclosporin is an immunosuppressant, with a synergistic and dual mechanism of action. By inhibiting calcineurin, voclosporin blocks IL-2 expression and T-cell mediated immune responses and stabilizes the podocyte in the kidney. It has been shown to have a more predictable pharmacokinetic and pharmacodynamic relationship (potentially requires no therapeutic drug monitoring), an increase in potency (vs cyclosporin), and an improved metabolic profile compared to legacy CNIs. Aurinia anticipates that upon regulatory approval, patent protection for voclosporin will be extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act and comparable laws in other countries and until April 2028 with anticipated pediatric extension. Further, the new Notice of Allowanceis expected to result in the issuance of a U.S. patent with a term extending to December 2037. If the FDA approves the use of voclosporin for LN and the label for such use follows the dosing protocol under the Notice of Allowance, the issuance of this patent will expand the scope of intellectual property protection for voclosporin to December 2037.
Forward-Looking Statements
Certain statements made in this press release may constitute forward-looking information within the meaning of applicable Canadian securities law and forward-looking statements within the meaning of applicable United States securities law. These forward-looking statements or information include but are not limited to statements or information with respect to: voclosporin being a transformative drug for the potential treatment of proteinuric kidney diseases, such as lupus nephritis, as well as a unique opportunity for the treatment of dry eye syndrome; results from the Company’s Phase 3 trial in lupus nephritis by the end of 2019; timing for regulatory approval and commercialization of voclosporin for use in lupus nephritis; patent protection for voclosporin being extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 and until April 2028 with an anticipated pediatric extension; and intellectual property protection for voclosporin being extended to December 2037 in respect of a patent anticipated to be issued in connection with a new Notice of Allowance. It is possible that such results or conclusions may change based on further analyses of these data. Words such as “anticipate”, “will”, “believe”, “estimate”, “expect”, “intend”, “target”, “plan”, “goals”, “objectives”, “may” and other similar words and expressions, identify forward-looking statements. We have made numerous assumptions about the forward-looking statements and information contained herein, including among other things, assumptions about: the costs and expenses associated with Aurinia’s clinical trials; Aurinia receiving approval from regulators to proceed with commercialization; Aurinia being able to complete its clinical trials in a timely fashion; Aurinia being able to extend its patents on terms acceptable to Aurinia; and the validity of our patents. Even though the management of Aurinia believes that the assumptions made, and the expectations represented by such statements or information are reasonable, there can be no assurance that the forward-looking information will prove to be accurate.
Forward-looking information by their nature are based on assumptions and involve known and unknown risks, uncertainties and other factors which may cause the actual results, performance or achievements of Aurinia to be materially different from any future results, performance or achievements expressed or implied by such forward-looking information. Should one or more of these risks and uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those described in forward-looking statements or information. Such risks, uncertainties and other factors include, among others, the following: difficulties, delays, or failures we may experience in the conduct of our AURORA clinical trial; difficulties we may experience in completing the development and commercialization of voclosporin; the market for the LN business may not be as estimated; and regulatory authorities not granting approval for use of voclosporin in a commercial manner, or not granting patents or extensions for patents at all or as Aurinia currently anticipates. Although we have attempted to identify factors that would cause actual actions, events or results to differ materially from those described in forward-looking statements and information, there may be other factors that cause actual results, performances, achievements or events to not be as anticipated, estimated or intended. Also, many of the factors are beyond our control. There can be no assurance that forward-looking statements or information will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, you should not place undue reliance on forward-looking statements or information.
Except as required by law, Aurinia will not update forward-looking information. All forward-looking information contained in this press release is qualified by this cautionary statement. Additional information related to Aurinia, including a detailed list of the risks and uncertainties affecting Aurinia and its business can be found in Aurinia’s most recent Annual Information Form available by accessing the Canadian Securities Administrators’ System for Electronic Document Analysis and Retrieval (SEDAR) website or the U.S. Securities and Exchange Commission’s Electronic Document Gathering and Retrieval System (EDGAR) website.
QUEBEC CITY, April 11, 2019 /CNW Telbec/ – Opsens Inc. (“Opsens” or the “Company”) (TSX: OPS) (OTCQX: OPSSF) today reported its results for the second quarter of 2019.
HIGHLIGHTS
Consolidated revenues totaled $7.9M in the second quarter of 2019 compared with $5.4M in the corresponding period in 2018, an increase of $2.5M or 46%;
Fractional Flow Reserve (“FFR”) revenues of $4.9M for the second quarter 2019 compared with $3.3M for the same period last year, an increase of 48%;
Appointment of Alan Milinazzo as Executive Chairman of the Board of Directors;
$8M credit agreement with CIBC Innovation Banking;
Opsens’ dPR welcome in the interventional cardiology community in Japan, EMEA and Canada.
GROWTH STRATEGY
Opsens’ revenues reached a record level, a result of the growth of its medical business lines. FFR revenues increased by 48% year over year. “These results reflect cardiologists’ acceptance of the OptoWire’s distinctive features and the optimization of our sales approach deployed in the past year,” said Louis Laflamme, President and Chief Executive Officer of Opsens. “We are also pleased to report that our dPR, designed to assess intracoronary pressure without the injection of drug-stimulating medication, is gaining traction in the market,” he added.
“We continue to focus on improvements processes in sales, production and on driving innovation to capitalize on the growing physiology measurement market in cardiology,” concluded Mr. Laflamme.
Opsens’ product sales reached $7.2M in the three-month period ended February 28, 2019 compared with $5.3M in the same period the previous year. This increase is mainly explained by an increase in FFR revenues compared with the same quarter the year before. In addition, the Company recorded non-recurring license revenue of $0.7M ($0.1M for the same quarter last year) for consolidated total revenues of $7.9M ($5.4M for last year) for the quarter.
Gross margin increased to $4.6M for the quarter ended February 28, 2019 from $2.8M in the same period last year. Non-recurring licensing revenues accounted for $0.6M of this increase.
Net loss totaled $0.4M for the three-month period ended February 28, 2019, compared with a net loss of $1.3M for the same period last year.
As of February 28, 2019, the Company had a cash position of $10.3M ($10.9M as of August 31, 2018).
About Opsens Inc.
Opsens Inc. focuses mainly on the measure of FFR and dPR in interventional cardiology. Opsens offers an advanced optical-based pressure guidewire that aims at improving the clinical outcome of patients with coronary artery disease. Its flagship product, the OptoWire, is a second-generation fiber optic pressure guidewire designed to provide the lowest drift in the industry and excellent lesions access. The OptoWire has been used in the diagnosis and treatment of over 60,000 patients in more than 30 countries. It is approved for sale in the United States, European Union, Japan, and Canada.
Opsens is also involved in industrial activities in developing, manufacturing and installing innovative fibre optic sensing solutions for critical applications.
Forward-looking statements contained in this press release involve known and unknown risks, uncertainties and other factors that may cause actual results, performance and achievements of Opsens to be materially different from any future results, performance or achievements expressed or implied by the said forward-looking statements.
Neither TSX nor its Regulation Services Provider (as that term is defined in the policies of the TSX) accepts responsibility for the adequacy or accuracy of this release.
For Additional Inquiries contact:
Louis Laflamme, CPA, CA Chief Executive Officer 418-781-0333
Robin Villeneuve, CPA, CA Chief Financial Officer 418-781-0333
Breakthrough medical platform offers vision of personalized tumor treatments with its novel sonic beam therapy
ANN ARBOR, MICHIGAN // Apr. 8, 2019 – HistoSonics, developer of a non-invasive robotics platform and novel beam therapy, announced today that it has closed a $54 million Series C financing. The round was led by Varian Medical Systems, Inc., the global leader in radiation therapy and oncology solutions, and included healthcare investors Johnson & Johnson Innovation – JJDC, Inc. (JJDC), Lumira Ventures, Venture Investors, the State of Wisconsin Investment Board, as well as participation from existing investors. The financing also included robotics pioneer Fred Moll, M.D., founder of Intuitive Surgical and Chairman and CEO of Auris Health. HistoSonics plans to use the proceeds of the financing to complete key regulatory and commercial milestones and expand development of its breakthrough platform.
“We believe that the HistoSonics platform offers a unique solution and significant promise to treat patients with a number of different diseases across global markets and care settings,” commented Greg Sorensen, Vice President of Strategy and Business Development at Varian and new HistoSonics Board of Directors member. Also joining the company’s Board as part of the financing are Gerry Brunk, Managing Director of Lumira Ventures, and an appointee from JJDC.
Above: HistoSonics’ non-invasive platform, Robotically Assisted Sonic Therapy (RAST)™
HistoSonics’ non-invasive platform, Robotically Assisted Sonic Therapy (RAST)™, combines advanced robotics and imaging with proprietary sensing technology to deliver personalized treatments with unparalleled precision and control, and uses the science of histotripsy and focused sound energy to generate pressures strong enough to liquify and completely destroy targeted tissues at sub-cellular levels. The company believes that the novel mechanism of action of their proprietary technology provides significant advantages to patients, including the ability of the treatment site to recover and heal quickly, as well as provides physicians the unique ability to monitor the destruction of tissue under continuous real-time visualization and control, unlike any modality that exists today.
“We are very excited to be adding this group of experienced investment partners who share in our vision and mission,” said President and CEO Mike Blue. “RAST™ will offer transformative change for both patients and physicians and will help overcome many of the major limitations and side effects of today’s cancer therapies. It has also shown great promise to work synergistically with other therapies and platforms, such as drug and immunotherapy, a big focus of our preclinical work, as well as with other surgical robotic platforms. We are confident that RAST™ will provide an entirely new experience for patients and physicians, as well as a more cost-effective alternative that better aligns with value-based healthcare initiatives, and we are thrilled to have such a strong syndicate joining us on this journey.”
The new financing follows a year of impressive progress at HistoSonics. The company achieved significant development milestones and generated compelling early clinical evidence, as well as continued to add deep domain experience and industry leadership to its team. HistoSonics was also recognized as one of the most promising start-ups with a top 10 ranking in The Observer’s “Top 20 Flyover Tech”.
“Venture Investors has been an investor in HistoSonics from the start,” added Jim Adox, Chairman of the Board “and it is incredibly exciting to see the progress we have made over the past several years. The vision of the company from the very beginning has been to develop a true platform that will revolutionize how patients are treated across a broad spectrum of diseases, including some of the most significant cancers, and it is very gratifying to now see the tremendous advancement of the system as it nears market launch.”
About HistoSonics
HistoSonics is a venture-backed medical device company developing a non-invasive robotic platform and novel beam therapy, Robotically Assisted Sonic Therapy (RAST)™ . RAST™ uses the science of histotripsy and the pressure created by focused sound energy to liquify and destroy targeted tissue, including diseased tissue and tumors, at sub-cellular levels. The company’s new platform deliverers personalized, tissue specific treatments with unparalleled precision and control, and without the undesirable side effects of many of today’s interventional and surgical modalities. Histotripsy was developed at the University of Michigan and exclusively licensed to HistoSonics. The company is led by a team of experienced domain experts and industry leaders with offices in Ann Arbor, Michigan and Minneapolis, MN.
Lumira Ventures is proud to sponsor the 6th Annual Health Innovation Week (#HIW2019) from April 1-5, 2019 presented by MaRS Discovery District and Founding Partner, TO Health!
Health Innovation Week (#HIW2019) is Canada’s largest gathering of health startups, investors and the health ecosystem and allows participants to network, secure funding and learn from industry experts. This year’s conference focuses on 3 themes: Innovation, Investment and Adoption.
As part of programming, Lumira Ventures will participate on April 2, 2019 as part of MaRS HealthKick Invest. MaRS HealthKick Invest brings together innovators in the therapeutic, diagnostic, medical device, digital health and consumer health spaces and connects them to global venture capital funds, family offices, angel investors and other capital partners interested in investing in the future of health.
April 2, 2019 – 9:30AM EST – Early Stage Digital Health & Medical Device Investing
Photo Credit: Laura Arsie Gerry Brunk, Managing Director, Lumira Ventures
Moderated by Sheryl Thingvold, Senior Advisor, Medical Devices at MaRS Health Ventures, Gerry Brunk, Managing Director at Lumira Ventures will participate in this panel discussion. Gerry will also participate as a judge in the HealthKick Bio-Pharma Pitch Competition at 1:30PM EST.
April 2, 2019 – 11:30AM EST – Understanding How VCs Are Motivated to Invest
Photo Credit: Laura Arsie Peter van der Velden, Managing General Partner, Lumira Ventures
Moderated by Ying Tam, MaRS Health Ventures, Peter van der Velden, Managing General Partner at Lumira Ventures will participate in this panel discussion.
April 2, 2019 – 3:00PM EST – Early Stage Therapeutics Investing
Photo Credit: Laura Arsie Jacki Jenuth, Partner, Lumira Ventures
Moderated by Parimal Natwani, VP, MaRS Innovation, Jacki Jenuth, Partner at Lumira Ventures, will participate in this discussion.
“The MAKO Robot is an essential tool that helps us in surgery when we do a joint replacement like total hips and total knees and partial total knees. For us in this area, we’re the first ones to provide this robotic arm technology to assist surgeons in making their cuts, and their alignments, perfect in every joint, and so what makes it distinct, is that no one else can offer this in the region. Robotics is really important as an assistant tool to the surgeon. The robot doesn’t replace the surgeon, but what it does is provide extra guidance and extra assistance to make sure that every single cut and every alignment is perfect every single time. The MAKO Robot is a really important step in the way we do total joint surgery.”
“Geinsinger has a long history of being at the forefront of technology and innovation. The MAKO Robot is just another small step in that direction. Our founder Abigail Geinsinger said ‘Make it the best’ and there’s no question in my mind that having this as an assistive technology makes our total joint care the best.”
“The ideal candidate would be anybody considering a total joint replacement of their hip or knee today. Because the MAKO robotic procedure is less innovative and allows us to make more accurate cuts, we believe it can lead to faster recovery and better outcomes.”
Michael Suk, MD, Chair of the Geisinger Musculoskeletal Institute and the Department of Orthopaedic Surgery
AVID100 is the only clinical-stage anti-EGFR ADC that targets both wild-type and mutant forms of EGFR with limited off-tumor toxicity due to novel mechanism of action
Phase 1 confirmed that AVID100 was well-tolerated and established a recommended phase 2 dose (RP2D) of 220 mg/m2 (~6 mg/kg) q3w, one of the highest amongst ADCs in development and predicted to be in therapeutic range
Phase 2 trials ongoing in EGFR-overexpressing (IHC 3+) squamous cell carcinoma of the head and neck (SCCHN) and squamous non-small cell lung cancer (sqNSCLC)
Austin, TX, and Montreal, QC (Mar. 29, 2019) – Forbius, a clinical-stage company that develops novel biologics for the treatment of fibrosis and cancer, announces today that it will report the results from its Phase 1 dose-finding trial with novel, tumor selective, anti-epidermal growth factor receptor (EGFR) antibody-drug conjugate (ADC) AVID100 at the AACR Annual Meeting 2019. An additional AACR poster presentation will feature preclinical data confirming AVID100’s novel mechanism of action, which potently targets tumor cells while sparing EGFR-expressing non-tumor cells.
Presentation Details:
CT056 / 13 – A Phase Ia/IIa trial of AVID100, an anti-EGFR antibody-drug conjugate
Poster presentation: April 1, 8:00 AM – 12:00 PM EST, Section 17 Author: N. Lakhani (Abstract available here)
AVID100 was well-tolerated in a Phase 1 dose-escalation study in patients with advanced solid tumors of epithelial origin (any EGFR status)
Most common treatment-related adverse events (TRAEs, in > 25% patients) were rash (66.7%; grade 1 or 2), nausea (41.7%; grade 1 or 2), and fatigue (29.2%; grade 1 or 2)
Recommended Phase 2 dose (RP2D) of 220 mg/m2 (~6 mg/kg) confirmed, one of the highest RP2Ds reported for maytansinoid payload ADCs (Deslandes, 2014)
Phase 2 (AVID100-01; NCT03094169) enrollment ongoing to evaluate AVID100 efficacy, safety, and tolerability in patients with EGFR-overexpressing (IHC 3+) SCCHN and sqNSCLC
218 / 9 – AVID100 is an anti-EGFR ADC that promotes DM1-meditated cytotoxicity on cancer cells but not on normal cells
Poster presentation: March 31, 1:00 PM – 5:00 PM EST, Section 9 Author: M. Thwaites (Abstract available here)
AVID100 is highly potent and selectively cytotoxic against EGFR-expressing cancer cells, while sparing normal EGFR-positive keratinocytes
Protection of EGFR-expressing normal cells is shown to be a function of AVID100’s antibody moiety, which inhibits EGFR signaling and proliferation in normal cells
About AVID100 and the AVID100-01 Trial
AVID100 is a highly potent EGFR-targeting antibody-drug conjugate (ADC) that was engineered to achieve enhanced anti-tumor efficacy without a corresponding increase in toxicity against skin and other EGFR-expressing normal tissues. In preclinical studies, AVID100 demonstrated significant anti-cancer activity, including in EGFR-overexpressing tumor models that are resistant to marketed EGFR inhibitors. AVID100 is the only anti-EGFR ADC in clinical development that targets both wild-type and mutant forms of EGFR.
In a successfully completed Phase 1 study, AVID100 reported a recommended Phase 2 dose (RP2D) of 220 mg/m2 (~6mg/kg), which is expected to be in the therapeutically active range based on preclinical efficacy studies. Treatment was generally well-tolerated, with the majority of treatment-related adverse events in the Phase 1 trial at RP2D being grade 1 or 2 in severity.
AVID100-01 (NCT03094169) is an open label, multi-center, dose-escalation study to evaluate the safety and efficacy of AVID100 in patients with confirmed EGFR-overexpressing tumors.
About Forbius
Forbius is a clinical-stage protein engineering company that designs and develops novel biologics for the treatment of fibrosis and cancer. Our current focus is the development of agents that target the transforming growth factor-beta (TGF-beta) and epidermal growth factor receptor (EGFR) pathways.
March 27, 2019 — Medexus Pharmaceuticals Inc (CVE:MDP) (FRA:P731) CEO Ken d’Entremont provides an overview of the company and highlights its recent financial reporting. The company is the results of the recent amalgamation of three specialty pharmaceutical companies: Pediapharm Inc, Medexus Inc, and Medac Pharma. As d’Entremont explains, the combined entity provides Medexus Pharmaceuticals with a footprint in both Canada and the United States, while expanding its therapeutic expertise to include pediatrics, autoimmune diseases, oncology, and rheumatology. The merger was undertaken to allow the company to rapidly scale. Further, Medexus Pharmaceuticals can now drive revenue while opex stays flat, benefiting from the organic growth of a larger product portfolio. Medexus Pharmaceuticals announced Q3 Fiscal 2019 revenues of $14.4 million, a 512 percent increase year-over-year.
About Medexus Pharmaceuticals Inc.
Medexus Pharmaceuticals Inc. is a leading specialty pharmaceutical company with a strong North American commercial platform. The Company’s vision is to provide the best healthcare products to healthcare professionals and patients, through our core values of Quality, Innovation, Customer Service and Teamwork. Medexus is focused on the therapeutic areas of auto-immune disease and pediatrics. The leading products are Rasuvo and Metoject, a unique formulation of methotrexate (auto-pen and pre-filled syringe) designed to treat rheumatoid arthritis and other auto-immune diseases; and Rupall, an innovative allergy medication with a unique mode of action.
For more information, please contact:
Ken d’Entremont, Chief Executive Officer Medexus Pharmaceuticals Inc. Tel.: 905-676-0003 E-mail: ken.dentremont@medexusinc.com
Roland Boivin, Chief Financial Officer Medexus Pharmaceuticals Inc. Tel.: 514-762-2626 ext. 202 E-mail: roland.boivin@medexusinc.com
VICTORIA, British Columbia (BUSINESS WIRE) — In recognition of World Kidney Day and National Kidney Month, Aurinia Pharmaceuticals Inc. (NASDAQ: AUPH/TSX: AUP), a late clinical-stage biopharmaceutical company with research ongoing in two kidney diseases, Lupus Nephritis (LN) and Focal Segmental Glomerulosclerosis (FSGS), today announced a host of activities, including an initiative with The National Kidney Foundation (NKF),designed to raise awareness for kidney disease. To find out more and to get involved in National Kidney Month, Aurinia invites all to:
Explore important resources and activities from NKF and ALL IN, Aurinia’s educational program and support community for people living with LN, on The ALL IN Program website. Aurinia is working with the NKF to go #ALLINorange throughout the month of March. ALL IN community members will receive #ALLINorange buttons to wear to show support for kidney disease, as well as receive access to helpful information. Learn more.
Visit our National Kidney Month calendar on Aurinia’s corporate website to get information about kidney disease and a quick overview of the important kidney awareness activities happening in March.
Re-share, like or comment on the useful resources and surprising facts about kidney disease that we are posting from our handle @AuriniaPharma on Twitter and Facebook
“National Kidney Month is so important because it is imperative that kidney health becomes a part of everyday conversations,” said Joseph Vassalotti, MD, Chief Medical Officer with the National Kidney Foundation. “Kidney diseases such as lupus nephritis and FSGS can be devastating for people living with those diseases as well as their friends, family and loved ones. For these reasons, we are excited to work with Aurinia to both spread awareness and provide helpful resources.”
“Both LN and FSGS are serious and often debilitating diseases for which there are currently no FDA or EMA approved therapies,” said Neil Solomons, M.D. Chief Medical Officer at Aurinia. “Additionally, LN and FSGS can lead to an end-stage renal disease requiring dialysis or a kidney transplant. In extreme cases, these diseases can even lead to death. Aurinia is committed to raising awareness around these kidney diseases and to developing innovative products to potentially help,” he added.
Aurinia Pharmaceuticals is a late clinical-stage biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are impacted by serious diseases with a high unmet medical need. The company is currently developing an investigational drug, for the treatment of Lupus Nephritis (LN), Focal Segmental Glomerulosclerosis (FSGS) and Dry Eye Syndrome. The company is headquartered in Victoria, British Columbia and focuses its development efforts globally.
About Kidney Disease
In the United States, 30 million adults are estimated to have chronic kidney disease and most aren’t aware of it. 1 in 3 American adults is at risk for chronic kidney disease. Risk factors for kidney disease include diabetes, high blood pressure, heart disease, obesity, and family history. People of African American, Hispanic, Native American, Asian or Pacific Islander descent are at increased risk for developing the disease. African Americans are 3 times more likely than whites, and Hispanics are nearly 1.5 times more likely than non-Hispanics to develop end-stage renal disease (kidney failure).1
About National Kidney Foundation
The National Kidney Foundation (NKF) is the largest, most comprehensive, and longstanding patient-centric organization dedicated to the awareness, prevention, and treatment of kidney disease in the U.S.
Forward-Looking Statements
Certain of the statements made in this presentation may constitute forward-looking information within the meaning of applicable Canadian securities law and forward-looking statements within the meaning of applicable U.S. securities law. These forward-looking statements or information include, but are not limited to statements or information with respect to the projected worth of the lupus nephritis (LN) market, that voclosporin is potentially a best-in-class calcineurin-inhibitor (CNI) with robust intellectual property protection and exclusivity and the likelihood of data exclusivity in major markets, the expectation that voclosporin will be the only CNI with a label for LN, the expected progress of the AURORA study; the anticipated commercial potential of voclosporin for the treatment of LN and FSGS.
When used in these marketing materials, the words “anticipate”, “will”, “believe”, “estimate”, “expect”, “intend”, “target”, “plan”, “goals”, “objectives”, “may” and other similar words and expressions, identify forward-looking statements or information.
We have made numerous assumptions about the forward-looking statements and information contained herein, including among other things, assumptions about: the market value for the LN program; that another company will not create a substantial competitive product for Aurinia’s LN business without violating Aurinia’s intellectual property rights; and the size of the LN market. Even though the management of Aurinia believes that the assumptions made and the expectations represented by such statements or information are reasonable, there can be no assurance that the forward-looking information will prove to be accurate.
Forward-looking information by their nature are based on assumptions and involve known and unknown risks, uncertainties and other factors which may cause the actual results, performance or achievements of Aurinia to be materially different from any future results, performance or achievements expressed or implied by such forward-looking information. Should one or more of these risks and uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those described in forward-looking statements or information. Such risks, uncertainties and other factors include, among others, the following: the market for the LN business may not be as estimated; and competitors may arise with similar products.
Although we have attempted to identify factors that would cause actual actions, events or results to differ materially from those described in forward-looking statements and information, there may be other factors that cause actual results, performances, achievements or events to not be as anticipated, estimated or intended. Also, many of the factors are beyond our control. There can be no assurance that forward-looking statements or information will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, you should not place undue reliance on forward-looking statements or information.
Except as required by law, Aurinia will not update forward-looking information. All forward-looking information contained in this presentation is qualified by this cautionary statement. Additional information related to Aurinia, including a detailed list of the risks and uncertainties affecting Aurinia and its business can be found in Aurinia’s most recent Annual Information Form available by accessing the Canadian Securities Administrators’ System for Electronic Document Analysis and Retrieval (SEDAR) website or the U.S. Securities and Exchange Commission’s Electronic Document Gathering and Retrieval System (EDGAR) website.
1st Spine PMA approved by the FDA in more than 3 years
15-month PMA approval process vs. 30-month average for Ortho/Spine in the last 10 years
WASHINGTON (PRNewswire) — On March 7, 2019, MCRA, LLC announced its role in the successful Premarket Approval (PMA) application decision by the U.S. Food and Drug Administration (FDA) to approve the Orthofix M6-C™ artificial cervical disc for the treatment of single level cervical disc degeneration. A next-generation artificial cervical disc, the M6-C disc developed by Spinal Kinetics LLC is the only artificial cervical disc designed to mimic the anatomic structure of a natural disc. To date, MCRA has assisted companies with 11 successful PMAs, including seven in the spine.
Spinal Kinetics, which was acquired by Orthofix Medical Inc. in 2018, initially retained MCRA, LLC in 2008 during the early stages of developing the clinical study strategy for the M6-C artificial cervical disc. The FDA approved the PMA for the M6-C disc on February 6, 2019, resulting in a 15-month review time between PMA submission and FDA approval. Additionally, the PMA approval for the M6-C artificial cervical disc is the first original PMA approval for a spinal device since 2015.
Tom Afzal, Founder and CEO of Spinal Kinetics and current General Manager of Motion Preservation at Orthofix, said, “We brought MCRA in early in our U.S. clinical trial process to evaluate our strategy and, thereafter, work with the team to support the PMA submission and ongoing FDA interactions. They provided tremendous support in analysis and presentation of data, input on responses, communications with the FDA, and overall messaging of safety and effectiveness of the M6-C artificial cervical disc. Their interactions with the FDA provided a high level of confidence for us and led to a timely PMA approval. They were an excellent partner and our collaboration with them couldn’t have gone better.”
Kevin B. McGowan, Director of Regulatory Affairs at MCRA, LLC added, “We are extremely pleased to have worked with Spinal Kinetics in clinical study strategy and ultimately through the FDA’s rigorous PMA review process to enable the company to provide U.S. patients with another treatment option for cervical disc replacement. The MCRA regulatory team remains committed to helping companies bring new medical products to the U.S. market through our scientific and clinical knowledge coupled with our proven experience with the FDA approval process.”
About MCRA, LLC
Founded in 2004, MCRA, LLC is a leading adviser and clinical research organization to the neuro-musculoskeletal and orthopedic industry. MCRA’s value lies in its industry experience and integration of five business value creators: regulatory, reimbursement, clinical research, healthcare compliance and quality assurance. MCRA’s integrated approach of these key value creating initiatives provides unparalleled expertise for its clients. MCRA has offices in Washington, DC, Manchester, CT, and New York, NY, and serves nearly 500 clients globally. MCRA has a demonstrated history of driving success in all areas of the medical device industry including spine, orthopedics, cardio-vascular, diagnostic imaging, endoscopy, ophthalmics, general/plastic surgery, drug delivery, wound care, diabetes, dental, general healthcare, nephrology, neurology, cardiology, and in vitro diagnostic (IVD) devices.
For Additional Information Contact:
David W. Lown General Manager, MCRA, LLC Phone: (212) 583-0250 ext. 2111 Email: dlown@mcra.com
SUMMIT, N.J. (PRNewswire) — Engage Therapeutics, Inc., a clinical-stage biopharmaceutical company developing a Rapid Epileptic Seizure Termination (REST) therapy for people who experience a predictable pattern of epileptic seizures, today announced it will present clinical data from the open-label portion of the randomized Phase 2b StATES Study (Staccato Alprazolam Terminates Epileptic Seizures) at the 2019 American Academy of Neurology Annual Meeting, which will be held May 4–10, 2019, in Philadelphia.
The poster is
entitled, “A Two-Part, Double-Blind, Placebo-Controlled, Inpatient,
Dose-Ranging Efficacy Study of Staccato Alprazolam (STAP-001) in Patients with
Epilepsy with a Predictable Seizure Pattern: Results from the Initial
Open-Label Feasibility Part.”
“The Open-Label data are encouraging and we are very pleased to have the opportunity to showcase research that could lay the groundwork for an ultra-rapid option for acute seizure rescue treatment,” said Dr. Jaqueline French, the study’s principal investigator and professor in the Department of Neurology at NYU Langone Health, in the Comprehensive Epilepsy Center, and founder/director of the Epilepsy Study Consortium. “Many patients with epilepsy live with the fear that a seizure may occur at any moment, highlighting the need for a rescue medication that could give patients the confidence that a seizure could be rapidly aborted.”
Presentation
details are as follows:
Presenter:
Jacqueline French, M.D.
Session Date and
Time:
May 5, 11:30 a.m. to 6:30 p.m. EDT
Poster
Presentation Number: 5-023
The StATES study will enroll patients from more than 45 trial sites in the U.S., Australia, Canada, and Jamaica. Approximately one hundred fifteen subjects will be enrolled in the double-blind, placebo-controlled portion of the trial which commenced on January 2019.
About Engage
Therapeutics, Inc.
Engage Therapeutics, Inc. is developing Staccato alprazolam, for the immediate cessation of active and acute epileptic seizures. The investigational product is in the Rapid Epileptic Seizure Termination (REST) category of products. It is a small, easy-to-use hand-held drug-device combination that leverages a fast-acting FDA-approved delivery system with FDA-approved alprazolam. Staccato alprazolam demonstrated reduction of seizure-like activity in a photosensitivity model in a Phase 2a proof of concept study. The product has now proceeded into a Phase 2b randomized study known as the StATES study. Engage Therapeutics is based in Summit, N.J.
IRVINE, Calif., — Edwards Lifesciences CorporationEW, -0.99% the global leader in patient-focused innovations for structural heart disease and critical care monitoring, today announced two strategic transactions involving companies with structural heart disease technologies.
Separately, Edwards has also acquired certain assets of Mitralign, Inc.,
including intellectual property and associated clinical and regulatory
experience. Mitralign is a developer of an investigational transcatheter
annuloplasty system designed to treat functional mitral and tricuspid
regurgitation.
Additional terms of these transactions remain confidential. Today’s
announcement is not expected to impact Edwards’ 2019 financial guidance.
About Edwards Lifesciences Edwards Lifesciences, based in Irvine, Calif., is the global leader in patient-focused medical innovations for structural heart disease, as well as critical care and surgical monitoring. Driven by a passion to help patients, the company collaborates with the world’s leading clinicians and researchers to address unmet healthcare needs, working to improve patient outcomes and enhance lives.
This news release includes forward-looking statements within the meaning of
Section 27A of the Securities Act of 1933 and Section 21E of the Securities
Exchange Act of 1934. These forward-looking statements include, but are not
limited to, statements regarding financial guidance, expected product benefits,
transaction expectations, and other statements that are not historical facts.
Forward-looking statements are based on estimates and assumptions made by
management of the company and are believed to be reasonable, though they are
inherently uncertain and difficult to predict. Our forward-looking statements
speak only as of the date on which they are made and we do not undertake any
obligation to update any forward-looking statement to reflect events or
circumstances after the date of the statement.
Forward-looking statements involve risks and uncertainties that could cause results to differ materially from those expressed or implied by the forward-looking statements based on a number of factors, including but not limited to, unexpected delays or changes in clinical trials, regulatory approvals and product introductions, unanticipated outcomes of longer-term clinical experience with the new products, or unanticipated manufacturing, legal, quality or regulatory delays or issues. These factors are detailed in the company’s filings with the Securities and Exchange Commission including its Annual Report on Form 10-K for the year ended December 31, 2018. These filings, along with important safety information about our products, may be found at edwards.com. Edwards, Edwards Lifesciences, and the stylized E logo are trademarks of Edwards Lifesciences Corporation. All other trademarks are the property of their respective owners. This statement is made on behalf of Edwards Lifesciences Corporation and its subsidiaries
LOS ANGELES, March 8, 2019 /CNW/ — Stellar Biotechnologies, Inc. (Nasdaq: SBOT), a leading manufacturer of a key protein utilized in immunotherapy and immuno-oncology development pipelines, has executed a share exchange agreement with privately-held Edesa Biotech Inc., a Canadian company, and Edesa’s shareholders to create a company focused on the development of innovative therapeutics for dermatological and gastrointestinal indications with clear unmet medical needs.
Under the terms of the share exchange agreement, Edesa shareholders have agreed to exchange their shares of Edesa for newly-issued common shares of Stellar. At the closing, Edesa will become a wholly-owned subsidiary of Stellar. Following the closing, current Stellar shareholders are expected to own approximately 10%, and the current shareholders of Edesa are expected to own approximately 90%, of the combined company on a fully-diluted basis, subject to a 2% upward or downward adjustment based upon the amount of Stellar’s working capital balance immediately prior to the closing. Following the closing, Stellar will change its name to “Edesa Biotech Inc.”
“We believe this proposed business combination provides new growth opportunities for Stellar shareholders. We have been impressed with Edesa’s management team and are looking forward to implementing a new vision for the combined company,” said Frank R. Oakes, Stellar President and Chief Executive Officer.
“This agreement marks another milestone for Edesa and our mission to efficiently develop novel, safe and effective treatments for conditions where patients have limited treatment options available,” said Par Nijhawan, MD, Chief Executive Officer of Edesa. “We believe we are at a significant inflection point in our company’s history and look forward to offering shareholders additional value creation opportunities as we reach milestones in our clinical programs.”
The proposed transaction, which will result in a change in control, is expected to close during the second quarter of 2019, subject to customary closing conditions, including Stellar shareholder approval for the issuance of Stellar common shares to acquire Edesa. Following closing, Stellar intends to develop a plan for the disposition of Stellar’s operations, which is expected to include the wind down or spin-off of Stellar’s legacy business. Following a diligent review of strategic alternatives, Stellar’s Board of Directors has determined that the share exchange agreement is fair and in the best interests of Stellar and Stellar’s shareholders. For further information about the proposed transaction, investors should refer to Stellar’s SEC filings.
H.C. Wainwright & Co. acted as transaction advisor and Greenburg Traurig LLP (USA) and McMillan LLP (Canada) acted as legal advisors to Stellar. Fasken Martineau DuMoulin LLP (Canada) and Stubbs Alderton & Markiles, LLP (USA) acted as legal advisors to Edesa.
About Stellar Biotechnologies Based north of Los Angeles at the Port of Hueneme, Stellar Biotechnologies, Inc. (Nasdaq: SBOT) is the leader in sustainable manufacture of Keyhole Limpet Hemocyanin (KLH), an immune-stimulating protein utilized as a carrier molecule in therapeutic vaccine pipelines (targeting cancers, immune disorders, Alzheimer’s and inflammatory diseases) and for assessing immune system function. KLH can also be used in immunotoxicology studies for monitoring the immunomodulatory effects of drug candidates. Stellar KLH is a trademark of Stellar Biotechnologies.
About Edesa Biotech Inc. Edesa Biotech Inc. is a clinical-stage company focused on efficiently developing innovative treatments that address significant unmet medical needs. The company’s leading product candidate, EB01, is a novel non-steroidal anti-inflammatory molecule for the treatment of allergic contact dermatitis which has demonstrated statistically significant improvements in multiple clinical studies. Edesa is based in Toronto, Canada.
Stellar Forward-Looking Statements This press release may contain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements may be identified by the use of words such as “anticipate,” “believe,” “plan,” “estimate,” “expect,” “intend,” “may,” “will,” “would,” “could,” “should,” “might,” “potential,” or “continue” and variations or similar expressions, including statements related to: the expectations related to the transaction, including the timing of the transaction, resulting ownership of the combined company, and the plans relating to the resulting business. Readers should not unduly rely on these forward-looking statements, which are not a guarantee of future performance. There can be no assurance that forward-looking statements will prove to be accurate, as all such forward-looking statements involve known and unknown risks, uncertainties and other factors which may cause actual results or future events to differ materially from the forward-looking statements. Such risks include, but may not be limited to: the possibility that Stellar may be unable to obtain shareholder approval required for the proposed transaction, the expected timing and likelihood of completion of the proposed transaction, the possibility that Stellar’s working capital decreases prior to the transaction, and therefore, the Stellar shareholders are subject to decreased ownership in the combined company, the inability to successfully integrate the businesses or the risk that such integration may be more difficult, time-consuming or costly than expected, the occurrence of any event, change or other circumstances that could give rise to the termination of the share exchange agreement, the inability of the parties to meet expectations regarding the accounting and tax treatments of the proposed transaction, the potential for the proposed transaction to involve unexpected costs, the risk that the parties may not be able to satisfy the conditions to the proposed transaction in a timely manner or at all, risks related to disruption of management time from ongoing business operations due to the proposed transaction, the risk that the expected benefits of the proposed combination are not realized, the risk that any announcements relating to the proposed transaction could have adverse effects on the market price of Stellar’s common stock, the ability to maintain and enforce patents and other intellectual property rights or the unexpected costs associated with such enforcement or litigation, as well as general economic and business conditions, technology changes, competition, changes in strategy or development plans, availability of funds and resources, anticipated requirements for operating capital, governmental regulations and the ability or failure to comply with governmental regulations, changes in trade policy and international law and other factors referenced in Stellar’s filings with securities regulators. Risks and uncertainties related to Edesa that may cause actual results to differ materially from those expressed or implied in any forward-looking statements include, but are not limited to: the ability of Edesa to obtain regulatory approval for or successfully commercialize any of its product candidates, the risk that access to sufficient capital to fund Edesa’s operations may not be available or may be available on terms that are not commercially favorable to Edesa, the risk that Edesa’s product candidates may not be effective against the diseases tested in its clinical trials, the risk that Edesa fails to comply with the terms of license agreements with third parties and as a result loses the right to use key intellectual property in its business, Edesa’s ability to protect its intellectual property and the timing and success of submission, acceptance and approval of regulatory filings. Many of these factors that will determine actual results are beyond Stellar’s, Edesa’s or the combined company’s ability to control or predict.
Other risks and uncertainties are more fully described in periodic filings with the SEC, including the factors described in the section entitled “Risk Factors” in our Annual Report on Form 10-K filed with the SEC on November 30, 2018 for the year ended September 30, 2018 and any Quarterly Reports on Form 10-Q filed thereafter, and in other filings that Stellar makes and will make with the SEC in connection with the proposed transactions, including the proxy statement described below under “Important Information and Where to Find It,” as well as its filings with the British Columbia Securities Commission. Existing and prospective investors are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. The statements made in this press release speak only as of the date stated herein, and subsequent events and developments may cause our expectations and beliefs to change. Unless otherwise required by applicable securities laws, we do not intend, nor do we undertake any obligation, to update or revise any forward-looking statements contained in this news release to reflect subsequent information, events, results or circumstances or otherwise.
Important Information and Where to Find It
Stellar and Edesa and certain of their directors and executive officers may become participants in solicitation of proxies from Stellar shareholders in connection with the proposed transaction. Additional Information regarding persons who may, under the rules of the SEC, be deemed to be participants in the solicitation of the Stellar shareholders in connection with the proposed transaction, and who have interests, whether as security holders, directors or employees of Stellar or Edesa or otherwise, which may be different from those of Stellar shareholders generally, will be provided in the proxy statement and other materials to be filed with the SEC.
Each member of Stellar’s board of directors and Stellar’s executive officers, and Edesa’s board of directors and Edesa’s executive officers may be deemed “participants” in the solicitation of proxies from the Stellar shareholders in connection with the proposed transaction.
Information regarding the special interests of these directors and executive officers in the transaction will be included in the proxy statement referred to above. Additional information regarding Stellar’s directors’ and executive officers’ respective interests in Stellar by security holdings or otherwise is set forth in Stellar’s Annual Report on Form 10-K for the year ended September 30, 2018, as filed with the SEC on November 30, 2018.
This communication does not constitute an offer to sell or the solicitation of an offer to buy any securities or a solicitation of any vote or approval. A definitive proxy statement and a proxy card will be filed with the SEC and will be mailed to Stellar’s shareholders seeking any required shareholder approvals in connection with the proposed transaction. BEFORE MAKING ANY VOTING OR INVESTMENT DECISION, INVESTORS AND SHAREHOLDERS ARE URGED TO READ THE PROXY STATEMENT (INCLUDING ANY AMENDMENTS OR SUPPLEMENTS THERETO) AND ANY OTHER RELEVANT DOCUMENTS THAT STELLAR MAY FILE WITH THE SEC WHEN THEY BECOME AVAILABLE BECAUSE THEY WILL CONTAIN IMPORTANT INFORMATION ABOUT THE PROPOSED TRANSACTION. Shareholders may obtain, free of charge, copies of the definitive proxy statement and any other documents filed by Stellar with the SEC in connection with the proposed transaction at the SEC’s website (http://www.sec.gov), at Stellar’s website or by writing to the Corporate Secretary at Stellar Biotechnologies, Inc., 332 E. Scott Street, Port Hueneme, California 93041.
Investor Relations
Gary Koppenjan, Stellar Biotechnologies, Inc. (805) 488-2800, ir@stellarbiotech.com
Michael Brooks, Edesa Biotech Inc. (905) 475-1234, investors@edesabiotech.com
This trial will evaluate the efficacy of AVID100 in SCCHN patients with EGFR IHC 3+ tumors
AVID100 is the most advanced, broadly active anti-EGFR ADC in clinical development
20% of SCCHN patients highly overexpress EGFR; no therapy is approved for these patients
Austin, TX, and Montreal, QC (Mar. 7, 2019) – Forbius, a clinical-stage company that develops novel biologics for the treatment of fibrosis and cancer, announced today that the first patient has been dosed in a Phase 2a squamous cell carcinoma of the head and neck (SCCHN) clinical trial.
The majority of SCCHN patients have tumors that overexpress epidermal growth factor receptor (EGFR) and approximately 20% have tumors that highly overexpress EGFR (more than 50% of cells with EGFR 3+ staining by a validated immunohistochemistry assay). No therapy is approved for the treatment of EGFR-overexpressing SCCHN.
AVID100 is a highly potent EGFR-targeting antibody-drug conjugate (ADC) engineered to achieve enhanced anti-tumor efficacy without a corresponding increase in toxicity against skin and other EGFR-expressing normal tissues. In preclinical studies, AVID100 demonstrated significant anti-cancer activity in EGFR overexpressing tumor models resistant to marketed EGFR inhibitors. AVID100 is the only broadly active anti-EGFR ADC in clinical development.
A recommended Phase 2 dose (RP2D) of 220 mg/m2 (~6mg/kg) was established for AVID100 in a completed Phase 1 study. This RP2D is expected to be in the therapeutically active range based on preclinical efficacy studies. The majority of treatment related adverse events in the Phase 1 trial at RP2D were well-tolerated and grade 1 or 2 in severity.
AVID100-01 is an open label, multicenter, dose-escalation study to evaluate the safety and efficacy of AVID100 in patients with confirmed EGFR-overexpressing tumors.
About Forbius
Forbius is a clinical-stage protein engineering company that designs and develops novel biologics for the treatment of fibrosis and cancer. Our current focus is on the development of agents that target the transforming growth factor-beta (TGF-beta) as well as the epidermal growth factor receptor (EGFR) pathways.
Alan Milinazzo, Partner, Global Healthcare and Life Sciences Practice – Heidrick & Struggles (PRNewsFoto/Heidrick & Struggles)
Quebec City, Quebec, March 4, 2019 – Opsens Inc. (“Opsens”) (TSX:OPS) (OTCQX:OPSSF) is pleased to announce the appointment of Mr. Alan Milinazzo as Executive Chairman of the Board of Directors.
“We are delighted to see Alan, a renowned leader in the medical device industry, join us. Alan has had extensive success in building high-performance commercial teams in interventional cardiology. Alan’s extensive experience as an executive of leading global businesses and his expertise in driving adoption of new technologies for interventional cardiology, spine, and surgical products will be a precious asset for Opsens,” said Denis M. Sirois who is transitioning his Chairmanship role to Mr. Milinazzo. Sirois continues, “Opsens will greatly benefit from Alan’s broad board experience and his outstanding track record of two decades of executive management in the healthcare industry”.
“We have ambitious goals and our growth is just beginning. We are fortunate to have Alan join our board and benefit from his deep knowledge of the North American and international interventional cardiology market, as we expand global marketing of our optical-based FFR and dPR products to assess intracoronary pressure and designed to provide the lowest drift in the industry,” said Louis Laflamme, President and CEO of Opsens.
“I am excited to be joining the Opsens board of directors at such a dynamic phase of the company’s growth and evolution. The company is well positioned to drive adoption of its state-of-the-art physiology and optical based technologies and to continue to bring important innovation to the minimally invasive markets,” said Mr. Milinazzo. “I am looking forward to working with the Board and Management to accelerate and optimize the market opportunities for our technologies and in doing so, to unlock significant value for our shareholders,” he added.
Alan Milinazzo is a Partner at Heidrick & Struggles’ in Boston and a member of the Global Healthcare and Life Sciences Practice specializing in the Medical Device sector. Prior to joining Heidrick & Struggles, Alan was Chief Executive Officer of InspireMD, a pioneer in embolic prevention systems (EPS) for coronary and vascular applications. He previously served as President and Chief Executive Officer of Orthofix International N.V., a $600 million publicly traded global orthopedic and Spine Company, as well as general manager of Medtronic, Inc.’s coronary and peripheral vascular businesses where he was instrumental in the development and commercialization of several key products, including the company’s first coronary drug-coated stent platform, Endeavor. Mr. Milinazzo also spent 12 years with Boston Scientific in multiple global sales and marketing leadership roles during a period of unprecedented top line growth in the cardiology franchise.
Alan currently serves as a director of Flexion Therapeutics (Nasdaq: FLXN), CasMed (Nasdaq: CASM) and the Musculoskeletal Transplant Foundation. Prior directorships included LDR Spine (Nasdaq LDRH acquired by Zimmer-Biomet) Medpace (acquired by PE sponsor Cinven), HET Systems (acquired by Covidien) and LumenR (acquired by Boston Scientific). He earned a bachelor’s degree, cum laude, from Boston College and interned at the White House, the US House of Representatives and the John F. Kennedy Library.
Denis M. Sirois remains on the BOD
“Opsens warmly thanks Denis M. Sirois for his contribution as he held the helm of the board for the past four years. We are grateful for his innovative ideas and strategic thinking that has allowed Opsens to make great strides,” added Louis Laflamme. “Denis will remain on the Board to continue building the Company,” Laflamme concluded.
About Opsens Inc.
Opsens focuses mainly on the measure of FFR and dPR in interventional cardiology. Opsens offers an advanced optical-based pressure guidewire that aims at improving the clinical outcome of patients with coronary artery disease. Its flagship product, the OptoWire, is a 2nd-generation fiber optic pressure guidewire designed to provide the lowest drift in the industry and excellent lesions access. The OptoWire has been used in the diagnosis and treatment of over 50,000 patients in more than 30 countries. It is approved for sale in the United States, European Union, Japan, and Canada.
Opsens is also involved in industrial activities in developing, manufacturing and installing innovative fibre optic sensing solutions for critical applications.
Forward-looking statements contained in this press release involve known and unknown risks, uncertainties and other factors that may cause actual results, performance and achievements of Opsens to be materially different from any future results, performance or achievements expressed or implied by the said forward-looking statements.
Neither TSX nor its Regulation Services Provider (as that term is defined in the policies of the TSX) accept responsibility for the adequacy or accuracy of this release.
-30-
For further information, please contact:
Louis Laflamme, CPA, CA, Chief Executive Officer, 418.781.0333
Robin Villeneuve, CPA, CA Chief Financial Officer, 418.781.0333
This trial will evaluate the safety and anti-fibrotic effects of AVID200 in diffuse cutaneous systemic sclerosis patients
AVID200 is a rationally designed, highly potent inhibitor of TGF-beta 1 & 3
TGF-beta 1 & 3 are the principal drivers of fibrosis in systemic sclerosis and other indications, supporting broad utility of AVID200
Austin, TX, and Montreal, QC (Mar. 4, 2019) – Forbius, a clinical-stage company that develops biologics for the treatment of fibrosis and cancer, announced today that the first patient has been dosed in a diffuse cutaneous systemic sclerosis (SSc) Phase 1b trial with AVID200, a rationally designed and highly potent inhibitor of TGF-beta 1 & 3.
“The basic defect in SSc and most other fibrotic diseases is increased TGF-beta signaling. Selective TGF-beta inhibition by AVID200 could rapidly reverse fibrosis, and I am keen to investigate the potential of AVID200 to transform the treatment of SSc,” commented Coordinating Principal Investigator Robert Lafyatis, M.D., Professor of Medicine, Medsger Professor and Director of the Scleroderma Center at the University of Pittsburgh Medical Center.
TGF-beta signaling is central to SSc pathogenesis (Lafyatis, 2014), and TGF-beta isoforms 1 & 3, but not 2, correlate positively with disease severity (O’Connor et al., 2018). AVID200 selectively neutralizes TGF-beta 1 & 3 with best-in-class pM potency, thus simultaneously neutralizing the principal pro-fibrotic TGF-beta isoforms and providing optimal efficacy.
“For decades, safe and potent neutralization of TGF-beta has been the holy grail for the treatment of fibrotic diseases. To achieve this, our team of pioneers in the TGF-beta field identified the two principal disease-driving TGF-beta isoforms and designed selective inhibitors that simultaneously neutralize these isoforms, while sparing the isoform that is critical for safety,” commented Mr. Ilia Tikhomirov, CEO of Forbius. “AVID200 has the potential to transform the treatment of many diseases and is the first of several new generation TGF-beta inhibitors being developed by Forbius.”
About Diffuse Cutaneous Systemic Sclerosis and the AVID200-01 Trial
SSc is a rare, severe, and progressively debilitating fibrotic disease that predominately affects women in mid-life. The 10-year survival rate of SSc patients is approximately 55%. No therapeutic is currently approved for the treatment of SSc, which affects an estimated 50,000 people in the U.S. alone.
AVID200-01 (NCT03831438) is a Phase 1 open-label, dose-escalation study to evaluate safety, pharmacokinetics, pharmacodynamics, and anti-fibrotic activity of AVID200 in patients with documented SSc.
About AVID200: TGF-beta 1 & 3 Inhibitor
AVID200 is a rationally designed, highly potent TGF-beta 1 & 3 inhibitor undergoing Phase 1 clinical testing in fibrosis and solid tumors. TGF-beta 1 & 3 are the principal disease-driving isoforms, while TGF-beta 2 is responsible for normal cardiac function and is a positive regulator of hematopoiesis. AVID200’s selectivity for TGF-beta 1 & 3 was designed to achieve optimal efficacy, while circumventing cardiac and other safety issues that have limited the applicability of older-generation, non-selective TGF-beta inhibitors. Therefore, AVID200 is positioned to be an effective and well-tolerated therapeutic in a variety of clinical settings.
About Forbius
Forbius is a clinical-stage protein engineering company that designs and develops biotherapeutics for the treatment of fibrosis and cancer. Our current focus is the development of agents that target the transforming growth factor-beta (TGF-beta) as well as the epidermal growth factor receptor (EGFR) pathways.
Allowed claims cover an individualized flat-dosed pharmacodynamic treatment protocol utilized in the AURA-LV study and the ongoing AURORA study in lupus nephritis
Claims have the potential to protect voclosporin’s method of use and dosing protocol for LN until December 2037
VICTORIA, British Columbia–(BUSINESS WIRE)– Aurinia Pharmaceuticals Inc. (NASDAQ:AUPH/ TSX:AUP) (the “Company” or “Aurinia”), a clinical-stage biopharmaceutical company focused on the global immunology market, today announced that it has received a Notice of Allowance from the United States Patent and Trademark Office (“USPTO”) for U.S. patent application 15/835,219, entitled “PROTOCOL FOR TREATMENT OF LUPUS NEPHRITIS”. The allowed claims broadly cover the novel voclosporin dosing protocol adhered to and required in both the previously reported Phase II AURA-LV study and the ongoing Phase III confirmatory AURORA study. Notably, the allowed claims cover a method of modifying the dose of voclosporin in patients with lupus nephritis (LN) based on patient-specific pharmacodynamic parameters.
This Notice of Allowance concludes a substantive examination of the
patent application at the USPTO, and after administrative processes are
completed and fees are paid, is expected to result in the issuance of a
U.S. patent with a term extending to December 2037. Issuance of the
patent will expand the scope of intellectual property protection for
voclosporin, which already includes robust manufacturing, formulation,
synthesis and composition of matter patents.
The Company has also filed for protection of this subject matter under
the Patent Cooperation Treaty (PCT) and has the option of applying for
similar protection in the member countries thereof. This may lead to the
granting of corresponding claims in the treaty countries which include
all the major global pharmaceutical markets. “These method of use claims
allowed in the U.S. broadly cover the personalized voclosporin dosing
protocol utilized across our LN program, which includes specific dose
modification requirements that we anticipate being incorporated into any
potential future label for voclosporin in LN,” said Michael R. Martin,
Chief Operating Officer of Aurinia.
“This Notice of Allowance is a significant milestone for Aurinia as it
enhances our current intellectual property portfolio and provides
potential exclusivity for Aurinia’s protocol for the treatment of
proteinuric kidney diseases, including LN, until late 2037. Importantly,
these claims provide validation of some unique and differentiating
features of voclosporin compared to the legacy CNIs.” stated Richard M.
Glickman, Chairman and CEO of Aurinia. “Establishing a robust
exclusivity platform is a critical part of our strategy as we work
towards regulatory approvals in the United States and internationally.”
About Aurinia
Aurinia Pharmaceuticals Inc. is a clinical-stage biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are suffering from serious diseases with a high unmet medical need. The company is currently developing voclosporin, an investigational drug, for the potential treatment of lupus nephritis (LN), focal segmental glomerulosclerosis (FSGS), and dry eye syndrome (DES). The company is headquartered in Victoria, British Columbia and focuses its development efforts globally.
About Voclosporin
Voclosporin, an investigational drug, is a novel and potentially best-in-class CNI with clinical data in over 2,400 patients across indications. Voclosporin is an immunosuppressant, with a synergistic and dual mechanism of action. By inhibiting calcineurin, voclosporin blocks IL-2 expression and T-cell mediated immune responses and stabilizes the podocyte in the kidney. It has been shown to have a more predictable pharmacokinetic and pharmacodynamic relationship (potentially requires no therapeutic drug monitoring), an increase in potency (vs cyclosporin), and an improved metabolic profile compared to legacy CNIs. Aurinia anticipates that upon regulatory approval, patent protection for voclosporin’s composition of matter will be extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act and comparable laws in other countries and until April 2028 with anticipated pediatric extension. Voclosporin’s unique dosing protocol used in both the AURA-LV and the AURORA studies for LN has also been granted a Notice of Allowance from the USPTO, these allowed claims have the potential to provide additional coverage for voclosporin until late 2037.
About Lupus Nephritis (LN)
LN in an inflammation of the kidney caused by Systemic Lupus Erythematosus (“SLE”) and represents a serious progression of SLE. SLE is a chronic, complex and often disabling disorder. The disease is highly heterogeneous, affecting a wide range of organs & tissue systems. Unlike SLE, LN has straightforward disease outcomes (measuring proteinuria) where an early response correlates with long-term outcomes. In patients with LN, renal damage results in proteinuria and/or hematuria and a decrease in renal function as evidenced by reduced estimated glomerular filtration rate (“eGFR”), and increased serum creatinine levels. LN is debilitating and costly and if poorly controlled, LN can lead to permanent and irreversible tissue damage within the kidney, resulting in end-stage renal disease (“ESRD”), thus making LN a serious and potentially life-threatening condition.
Forward-Looking Statements
Certain statements made in this press release may constitute forward-looking information within the meaning of applicable Canadian securities law and forward-looking statements within the meaning of applicable United States securities law. These forward-looking statements or information include but are not limited to statements or information with respect to the USPTO granting a new patent for the Company’s protocol with LN; the new patent having a patent term extending to 2037; Aurinia’s new patent claims being listed in the FDA’s Orange book; filings with the PCT leading to the granting of corresponding claims in treaty countries; voclosporin being potentially a best-in-class CNI with robust intellectual property exclusivity; the patent life for Aurinia’s patents; and the potential to extend that patent life on the occurrence of certain events. It is possible that such results or conclusions may change based on further analyses of these data. Words such as “anticipate”, “will”, “believe”, “estimate”, “expect”, “intend”, “target”, “plan”, “goals”, “objectives”, “may” and other similar words and expressions, identify forward-looking statements. We have made numerous assumptions about the forward-looking statements and information contained herein, including among other things, assumptions about: that another company will not create a substantial competitive product without violating Aurinia’s intellectual property rights; that the FDA will grant Aurinia approval for use of voclosporin with LN; that voclosporin in LN would qualify for publication in the FDA’s Orange Book; that the USPTO will issue a new patent once applicable steps have been followed and fees paid in respect of the Notice of Allowance; and Aurinia being able to extend its patents on terms acceptable to Aurinia. Even though the management of Aurinia believes that the assumptions made, and the expectations represented by such statements or information are reasonable, there can be no assurance that the forward-looking information will prove to be accurate.
Forward-looking information by their nature are based on assumptions and involve known and unknown risks, uncertainties and other factors which may cause the actual results, performance or achievements of Aurinia to be materially different from any future results, performance or achievements expressed or implied by such forward-looking information. Should one or more of these risks and uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those described in forward-looking statements or information. Such risks, uncertainties and other factors include, among others, the following: difficulties, delays, or failures we may experience in the conduct of our AURORA clinical trial; the FDA may not approve voclosporin for use with LN or for any other purpose; Aurinia not being able to extend or protect its patent portfolio for voclosporin or VOS; and competitors may arise with similar or more competitive products. Although we have attempted to identify factors that would cause actual actions, events or results to differ materially from those described in forward-looking statements and information, there may be other factors that cause actual results, performances, achievements or events to not be as anticipated, estimated or intended. Also, many of the factors are beyond our control. There can be no assurance that forward-looking statements or information will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, you should not place undue reliance on forward-looking statements or information.
Except as required by law, Aurinia will not update forward-looking information. All forward-looking information contained in this press release is qualified by this cautionary statement. Additional information related to Aurinia, including a detailed list of the risks and uncertainties affecting Aurinia and its business can be found in Aurinia’s most recent Annual Information Form available by accessing the Canadian Securities Administrators’ System for Electronic Document Analysis and Retrieval (SEDAR) website at www.sedar.com or the U.S. Securities and Exchange Commission’s Electronic Document Gathering and Retrieval System (EDGAR) website at www.sec.gov/edgar.
MPN-RC sponsored, NIH-supported Phase 1/2 trial in myelofibrosis to commence imminently
TGF-beta is a central driver of bone marrow fibrosis in myelofibrosis
AVID200 selectively inhibits TGF-beta 1 & 3, the principal fibrotic TGF-beta isoforms, while sparing TGF-beta 2, a positive regulator of hematopoiesis
Austin, TX and Montreal, QC (Feb. 14, 2019) – Forbius, a clinical-stage company that develops biologics for the treatment of cancer and fibrosis, announced today a collaboration agreement with the Icahn School of Medicine at Mount Sinai and the Myeloproliferative Neoplasm Research Consortium (MPN-RC). This collaboration launches an investigator-initiated trial (IIT) evaluating AVID200, a highly potent and isoform-selective TGF-beta inhibitor, as a potential treatment for myelofibrosis (MF). The Phase 1/2 clinical trial will be sponsored by the MPN-RC with NIH grant support and is expected to start during Q1 2019.
“We have demonstrated that blocking TGF-beta signaling reverses bone marrow fibrosis and restores hematopoiesis in preclinical models. We believe that selective TGF-beta inhibition by AVID200 could address the underlying cause of bone marrow failure and become the first disease-modifying therapy in MF. Our consortium is eager to commence evaluation of AVID200 in the upcoming clinical study,” commented Dr. Ronald Hoffman, founder of the MPN-RC and Director of the Myeloproliferative Disorders Research Program at the Icahn School of Medicine at Mount Sinai.
Forbius is evaluating the immuno-oncology and anti-fibrotic effects of AVID200 in Phase 1 trials, including solid tumors and systemic sclerosis. Additionally, Forbius is expanding the AVID200 clinical development program by supporting IITs through the provision of drug, scientific input, and collaboration on the conduct of translational studies. Additional details pertaining to the prospective IIT in MF will be disclosed in due course.
About the Myeloproliferative Neoplasm Research Consortium (MPN-RC)
The MPN-RC was founded in 2006 and is the only independent, multi-center, international consortium of scientists and clinicians that is dedicated to developing novel therapeutic strategies for MF and other myeloproliferative neoplasms (MPN). The MPN-RC is funded by the NIH to conduct clinical trials based on the most promising preclinical MPN research. The goal of the consortium is to adapt quickly in response to scientific advances and a changing clinical landscape, in order to develop effective therapeutics for MPN patients.
About AVID200
AVID200 is an isoform-selective and highly potent inhibitor of TGF-beta 1 & 3, the two principal pro-fibrotic TGF-beta isoforms. These TGF-beta isoforms are central regulators in the pathogenesis and progression of fibrotic diseases, including MF (Chagraoui et al., 2002). AVID200 was rationally designed to be minimally active against TGF-beta 2, which is a promoter of hematopoiesis and normal cardiac function. This optimal selectivity positions AVID200 to be an effective and well-tolerated therapeutic in MF and other fibrotic diseases.
About Myelofibrosis (MF)
MF is a rare, life-threatening blood cancer characterized by progressive bone marrow fibrosis, which causes ineffective hematopoiesis. Approximately 30,000 people in the US alone are affected by this disease. Currently, there are no approved therapies targeting the underlying bone marrow fibrosis available to MF patients.
About Forbius
Forbius is a clinical-stage protein engineering company that designs and develops biotherapeutics for the treatment of fibrosis and cancer. Our current focus is the development of agents targeting the transforming growth factor-beta (TGF-beta) and epidermal growth factor receptor (EGFR) pathways.
VOS showed statistical superiority to Restasis® on FDA-accepted objective signs of DES
42.9% of VOS subjects vs 18.4% of Restasis® subjects (p=.0055) demonstrated ≥ 10mm improvement in STT at Week 4
Primary endpoint of drop discomfort at 1-minute on Day 1 showed no statistical difference between VOS and Restasis®, as both exhibited low drop discomfort scores
Aurinia to advance VOS for the treatment of DES
VICTORIA, British Columbia–(BUSINESS WIRE)– Aurinia Pharmaceuticals Inc. (NASDAQ:AUPH/TSX:AUP), a clinical stage biopharmaceutical company focused on the global immunology market, today announced positive results for its exploratory Phase 2 head-to-head study evaluating the efficacy, safety and tolerability of voclosporin ophthalmic solution (VOS 0.2%) versus Restasis®(cyclosporine ophthalmic emulsion 0.05%) for the treatment of dry eye syndrome (DES). Both drugs were shown to be well-tolerated and there was no statistical difference between VOS and Restasis® for the primary endpoint as both drugs exhibited low drop discomfort scores.
On the key pre-specified secondary endpoints of Schirmer Tear Test/STT (an objective measure of tear production), and Fluorescein Corneal Staining/FCS (an objective measure of structural damage to the cornea), which are FDA-accepted efficacy endpoints, VOS showed rapid and statistically significant improvements over Restasis® at Week 4 (STT: p=.0051; FCS: p=.0003).
This 100-patient, double-masked, head-to-head study was designed to evaluate the efficacy, safety and tolerability of VOS versus Restasis® in subjects with DES. Both arms of the study received either VOS or Restasis® (1:1) administered twice daily, in both eyes, for 28 days. Key pre-specified secondary endpoints, which are FDA-accepted endpoints, include STT, FCS, and assessments of dry eye symptoms.
4-Week Pre-Specified Efficacy Endpoints (Signs)*
VOS
Restasis®
p-value vs. Restasis®
Schirmer Tear Test (STT)
(mm LS mean increase from baseline)
8.6
3.3
.0051
% of subjects showing ≥ 10mm improvement in STT
(basis of FDA approval for other CNIs and an improvement is considered to be clinically significant)
42.9%
18.4%
.0055
Fluorescein Corneal Staining (FCS)
(reduction in staining is clinically significant)
-2.2
-0.2
.0003
*worst eye
Both treatment arms also demonstrated substantial and statistically significant improvements on the Symptom Assessment in Dry Eye (SANDE) score from baseline to Week 4.
No serious adverse events were reported in the study, and there were no unexpected safety signals.
“Improvements in STT and FCS are considered by regulators to be two of the most clinically meaningful measures of efficacy in this disease. The rapid onset and overall efficacy (as measured by the STT and FCS) demonstrated by VOS in this head-to-head study conducted against Restasis® is astounding and could be a game changer in the treatment landscape for dry eye,” said Joseph Tauber, M.D., Principal Investigator and head of the renowned Tauber Eye Institute in Kansas City, MO.
Neil Solomons, M.D., Aurinia’s Chief Medical Officer said, “We are extraordinarily excited with the superior efficacy shown by VOS when compared to Restasis®, which is the current market leader for the treatment of DES in the US. The efficacy endpoints exceeded our expectations and provide further validation of the potential of VOS to provide a highly differentiated and efficacious treatment option for the more than 16 million patients living with this all-too-common disease.”
“Based on these positive data, we plan to aggressively advance VOS for the treatment of DES, which we believe can create considerable value for both patients and our shareholders,” said Richard M. Glickman, Chairman and CEO of Aurinia. “Our pursuit of further development of VOS provides the company with an enhanced pipeline that further capitalizes on the differentiating features of voclosporin and positions us for substantial growth.”
Aurinia will present the results of the clinical trial during a conference call and webcast presentation to be held at 8:00am ET Tuesday, January 22, 2019. A link to the live webcast and slides will be available on the Investors section of the Company’s website.
About Aurinia Aurinia Pharmaceuticals Inc. is a clinical stage biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are suffering from serious diseases with a high unmet medical need. The company is currently developing voclosporin, an investigational drug, for the potential treatment of lupus nephritis (LN), focal segmental glomerulosclerosis (FSGS), and dry eye syndrome (DES). The company is headquartered in Victoria, British Columbia and focuses its development efforts globally.
About VOS
VOS (voclosporin ophthalmic solution) is an aqueous, preservative free nanomicellar solution containing 0.2% voclosporin intended for use in the treatment of DES. Studies have been completed in rabbit and dog models, and a single Phase I has also been completed in healthy volunteers and patients with DES. VOS has IP protection until 2031.
About Dry Eye Syndrome (DES)
Dry eye syndrome (DES) is a chronic disease and is characterized by irritation and inflammation that occurs when the eye’s tear film is compromised by reduced tear production, imbalanced tear composition, or excessive tear evaporation. The impact of DES ranges from subtle, yet constant eye irritation to significant inflammation and scarring of the eye’s surface. Discomfort and pain resulting from DES can reduce quality of life and cause difficulty reading, driving, using computers and performing daily activities. While there are FDA approved therapies available for the treatment of DES, there is opportunity for potential improvement in the efficacy in addition to other measures such as onset of action, tolerability and dosing.
About Voclosporin Voclosporin, an investigational drug, is a novel and potentially best-in-class CNI with clinical data in over 2,400 patients across indications. Voclosporin is an immunosuppressant, with a synergistic and dual mechanism of action. By inhibiting calcineurin, voclosporin blocks IL-2 expression and T-cell mediated immune responses and stabilizes the podocyte in the kidney. It has been shown to have a more predictable pharmacokinetic and pharmacodynamic relationship (potentially requires no therapeutic drug monitoring), an increase in potency (vs cyclosporin), and an improved metabolic profile compared to legacy CNIs. Aurinia anticipates that upon regulatory approval, patent protection for voclosporin will be extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act and comparable laws in other countries and until April 2028 with anticipated pediatric extension.
About Restasis® RESTASIS® and RESTASIS MULTIDOSE™ Ophthalmic Emulsion help increase your eyes’ natural ability to produce tears, which may be reduced by inflammation due to Chronic Dry Eye. RESTASIS® and RESTASIS MULTIDOSE™ did not increase tear production in patients using anti-inflammatory eye drops or tear duct plugs.
Forward-Looking Statements
Certain statements made in this press release may constitute forward-looking information within the meaning of applicable Canadian securities law and forward-looking statements within the meaning of applicable United States securities law. These forward-looking statements or information include but are not limited to statements or information with respect to: Voclosporin Ophthalmic Solution (VOS) and the data as it relates to the phase 2 study: voclosporin being potentially a best-in-class CNI with robust intellectual property exclusivity, the development of VOS creating considerable value for patients and Aurinia’s shareholders; Aurinia being positioned for substantial growth; there being opportunities for improvement of efficacy, onset of action, tolerability and closing in DES; VOS being a game changer in the treatment landscape for dry eye; efficacy findings of VOS; the patent life for Aurinia’s patents; and the potential to extend that patent life on the occurrence of certain events. It is possible that such results or conclusions may change based on further analyses of these data. Words such as “anticipate”, “will”, “believe”, “estimate”, “expect”, “intend”, “target”, “plan”, “goals”, “objectives”, “may” and other similar words and expressions, identify forward-looking statements. We have made numerous assumptions about the forward-looking statements and information contained herein, including among other things, assumptions about: that another company will not create a substantial competitive product for Aurinia’s LN or DES business without violating Aurinia’s intellectual property rights; the burn rate of Aurinia’s cash for operations; the costs and expenses associated with Aurinia’s clinical trials; the planned studies achieving positive results; Aurinia being able to extend its patents on terms acceptable to Aurinia; and the size of the LN or DES market. Even though the management of Aurinia believes that the assumptions made, and the expectations represented by such statements or information are reasonable, there can be no assurance that the forward-looking information will prove to be accurate.
Forward-looking information by their nature are based on assumptions and involve known and unknown risks, uncertainties and other factors which may cause the actual results, performance or achievements of Aurinia to be materially different from any future results, performance or achievements expressed or implied by such forward-looking information. Should one or more of these risks and uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those described in forward-looking statements or information. Such risks, uncertainties and other factors include, among others, the following: difficulties, delays, or failures we may experience in the conduct of our AURORA, FSGS or DES clinical trials; difficulties we may experience in completing the development and commercialization of voclosporin or VOS; the market for the LN or DES business may not be as estimated; Aurinia may have to pay unanticipated expenses; estimated costs for clinical trials may be underestimated, resulting in Aurinia having to make additional expenditures to achieve its current goals; Aurinia not being able to extend or protect its patent portfolio for voclosporin or VOS; and competitors may arise with similar or more competitive products. Although we have attempted to identify factors that would cause actual actions, events or results to differ materially from those described in forward-looking statements and information, there may be other factors that cause actual results, performances, achievements or events to not be as anticipated, estimated or intended. Also, many of the factors are beyond our control. There can be no assurance that forward-looking statements or information will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, you should not place undue reliance on forward-looking statements or information.
Except as required by law, Aurinia will not update forward-looking information. All forward-looking information contained in this press release is qualified by this cautionary statement. Additional information related to Aurinia, including a detailed list of the risks and uncertainties affecting Aurinia and its business can be found in Aurinia’s most recent Annual Information Form available by accessing the Canadian Securities Administrators’ System for Electronic Document Analysis and Retrieval (SEDAR) website or the U.S. Securities and Exchange Commission’s Electronic Document Gathering and Retrieval System (EDGAR) website.
VANCOUVER, British Columbia — Zymeworks Inc. (NYSE/TSX: ZYME), a clinical-stage biopharmaceutical company developing multifunctional therapeutics, today reported the achievement of a new development milestone in its collaboration with Eli Lilly and Company (“Lilly”). In accordance with Zymeworks’ 2014 licensing and collaboration agreement with Lilly, Zymeworks will receive a milestone payment of US$8.0 million for Lilly’s submission of an IND application for an immuno-oncology bispecific antibody enabled by Zymeworks’ proprietary Azymetric™ platform.
“We believe that Lilly’s submission of a second IND on an Azymetric™ bispecific drug candidate within a six-month period is further evidence that Zymeworks’ platform technologies are enabling our global pharmaceutical partners to accelerate drug development into the clinic. As one of our first collaborators, Lilly’s successes demonstrate the ease of use, productivity and promise of our proprietary platforms, especially for advancing therapeutic programs into the clinical setting.”
– Ali Tehrani, Ph.D., President & CEO of Zymeworks.
Under its two licensing and collaboration agreements with Lilly, Zymeworks has granted Lilly worldwide licenses to research, develop, and commercialize multiple bispecific therapeutics directed towards Lilly’s targets. To date, Zymeworks has received multiple equity investments, an upfront licensing payment and multiple research and development milestone payments under these agreements. Zymeworks is also eligible to receive further development and commercial milestone payments and tiered royalties on global product sales.
About the Azymetric™ Platform
The Azymetric platform enables the transformation of monospecific antibodies into bispecific antibodies, giving the antibodies the ability to simultaneously bind two different targets. This unique technology enables the development of multifunctional biotherapeutics that can block multiple signaling pathways, recruit immune cells to tumors, enhance receptor clustering degradation, and increase tumor-specific targeting. These features are intended to enhance efficacy while reducing toxicities and the potential for drug resistance. Azymetric bispecifics have been engineered to retain the desirable drug-like qualities of naturally occurring antibodies, including low immunogenicity, long half-life and high stability. In addition, they are compatible with standard manufacturing processes with high yields and purity, potentially significantly reducing drug development costs and timelines.
About Zymeworks Inc.
Zymeworks is a clinical-stage biopharmaceutical company dedicated to the discovery, development, and commercialization of next-generation multifunctional biotherapeutics. Zymeworks’ suite of complementary therapeutic platforms and its fully-integrated drug development engine provide the flexibility and compatibility to precisely engineer and develop highly-differentiated product candidates. Zymeworks’ lead product candidate, ZW25, is a novel bispecific antibody currently being evaluated in an adaptive Phase 1 clinical trial. An Investigational New Drug (IND) application was recently accepted by the Food and Drug Administration (FDA) for its second product candidate, ZW49, a novel bispecific antibody-drug conjugate (ADC). Zymeworks is also advancing a deep pipeline of preclinical product candidates and discovery-stage programs in immuno-oncology and other therapeutic areas. In addition to Zymeworks’ wholly-owned pipeline, its therapeutic platforms have been further leveraged through multiple strategic partnerships with global biopharmaceutical companies.
This press release includes “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of Canadian securities laws, or collectively, forward-looking statements. Forward-looking statements in this news release include, but are not limited to, statements that relate to Zymeworks receipt of future milestone payments and royalties from Lilly or any of its other partners, the speed and success of drug discovery and development, the likelihood of Zymeworks’ partners advancing drug candidates to clinical trials and other information that is not historical information. When used herein, words and phrases such as “will,” “eligible to,” and similar expressions are intended to identify forward-looking statements. In addition, any statements or information that refer to expectations, beliefs, plans, projections, objectives, performance or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking. All forward-looking statements are based upon Zymeworks’ current expectations and various assumptions. Zymeworks believes there is a reasonable basis for its expectations and beliefs, but they are inherently uncertain. Zymeworks may not realize its expectations, and its beliefs may not prove correct. Actual results could differ materially from those described or implied by such forward-looking statements as a result of various factors, including, without limitation, market conditions and the factors described under “Risk Factors” in Zymeworks’ Quarterly Report on Form 10-Q for its fiscal quarter ended September 30, 2018 (a copy of which may be obtained at www.sec.gov and www.sedar.com). Consequently, forward-looking statements should be regarded solely as Zymeworks’ current plans, estimates, and beliefs. Investors should not place undue reliance on forward-looking statements. Zymeworks cannot guarantee future results, events, levels of activity, performance or achievements. Zymeworks does not undertake and specifically declines any obligation to update, republish, or revise any forward-looking statements to reflect new information, future events or circumstances or to reflect the occurrences of unanticipated events, except as may be required by law.