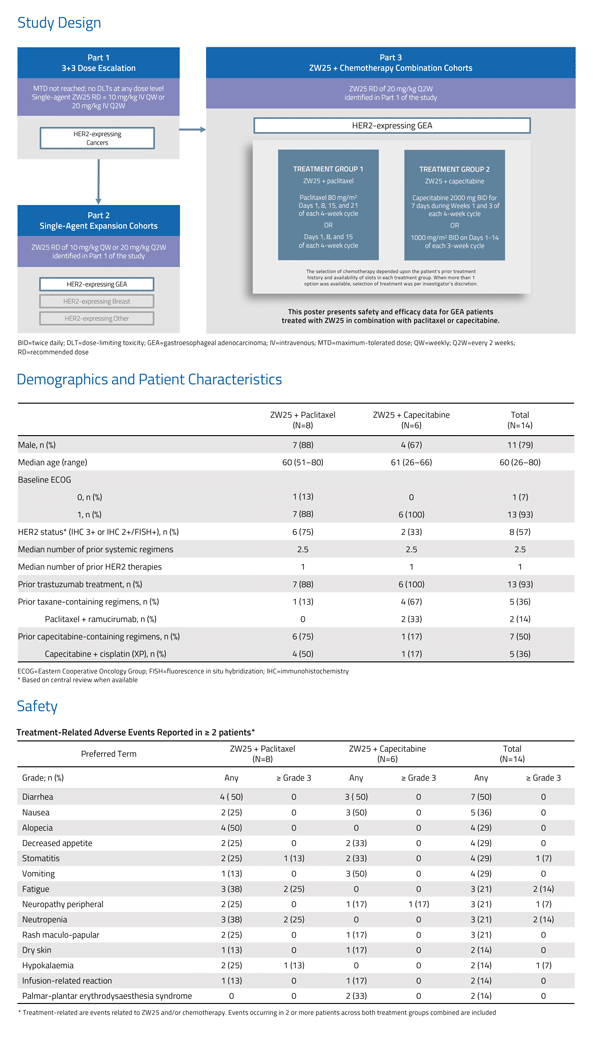
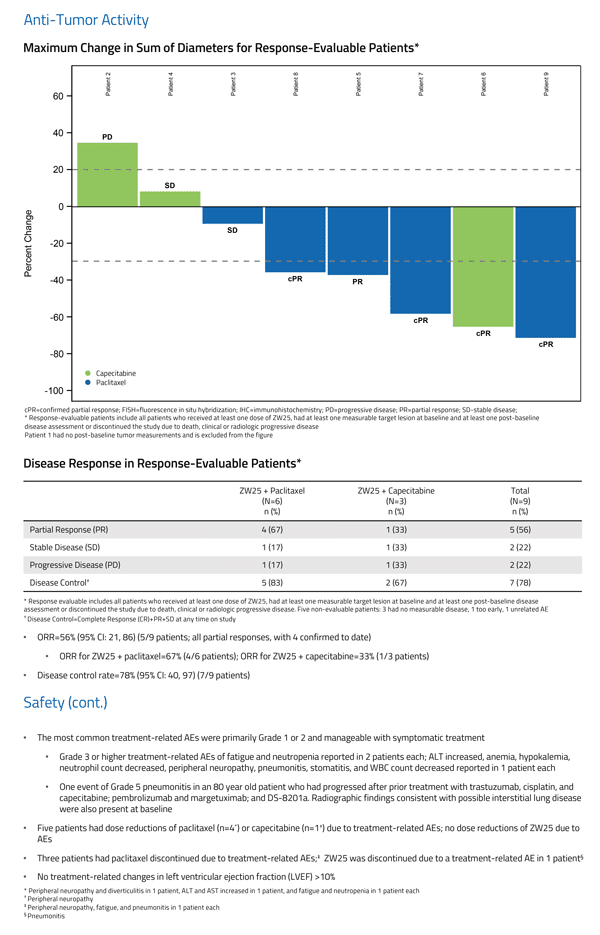
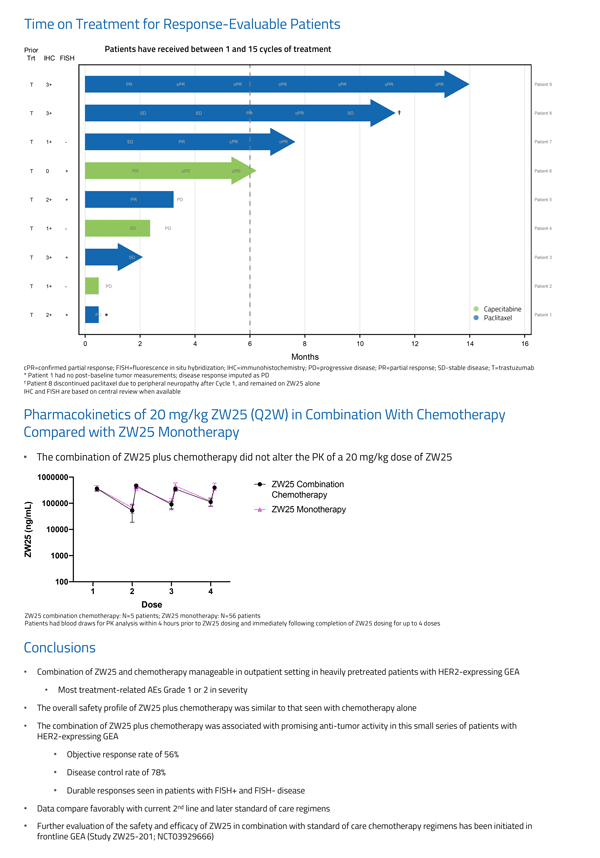
Related Article: Aurinia Reports First Quarter 2019 Financial Results & Recent Operational Highlights
Review condensed consolidated financials here: https://ir.auriniapharma.com/press-releases/detail/153
VICTORIA, British Columbia–(BUSINESS WIRE)– Aurinia Pharmaceuticals Inc. (NASDAQ: AUPH / TSX:AUP) (“Aurinia” or the “Company”), a late-stage clinical biopharmaceutical company focused on advancing voclosporin in multiple indications, today reported financial results for the three and six months ended June 30, 2019 and recent operational highlights. Amounts, unless specified otherwise, are expressed in U.S. dollars.
“During the past quarter, the Aurinia team has taken multiple steps forward preparing the organization for the next phase of its evolution into a commercial-stage entity. In anticipation of the AURORA Phase 3 results in lupus nephritis, we are appropriately scaling the organization ahead of data, which if confirmatory, sets the stage for a NDA filing during the first half of next year. If approved, we project a commercial launch of voclosporin in early 2021 as a potentially first-line treatment, in combination with mycophenolate mofetil and low-dose steroids for lupus nephritis,”
– Peter Greenleaf, President and Chief Executive Officer
Michael Martin, Chief Operating Officer stated, “In parallel, we are taking all of the steps necessary to initiate a Phase 2/3 clinical trial with VOS for the potential treatment of DES. This Phase 2/3 trial follows a well-established development path for ophthalmic drugs in this therapeutic area.”
Mr. Greenleaf further said, “With respect to our ongoing proof-of-concept study for FSGS, we have added additional clinical trial sites and have modified the study protocol to broaden the inclusion of patients who have received limited corticosteroids exposure prior to enrollment. We expect that these actions will lead to enhanced enrollment.”
VOS Phase 2/3 AUDREY (“Aurinia Dry Eye Study”) Clinical Study in DES
Based upon the VOS exploratory Phase 2a results, Aurinia has initiated plans for a Phase 2/3 study which is expected to enroll its first patient in the fourth quarter of 2019. This study will include certain critical regulatory requirements that the FDA has traditionally required for DES product approval, which include both dose-optimization along with a comparison to vehicle.
The Phase 2/3 clinical study otherwise known as, “THE AUDREY STUDY” is designed as a randomized, double-masked, vehicle-controlled, dose-ranging study to be conducted in the U.S., AUDREY will evaluate the efficacy and safety of VOS in subjects with DES. Approximately 480 subjects are to be enrolled. The study will consist of four arms and in a 1:1:1:1 randomization schedule to either 0.2% VOS, 0.1% VOS, 0.05% VOS or vehicle, dosed twice daily for 12 weeks. The primary outcome measure for the study is the proportion of subjects with 10mm improvement in Schirmer Tear Test (“STT”) at 4 weeks.
Secondary outcome measures, which will be assessed at multiple time points include, STT, Fluorescein Corneal Staining (“FCS”), change in eye dryness, burning/stinging, itching, photophobia, eye pain, and foreign body sensation, change in Symptom Assessment in Dry Eye (“SANDE”) score and additional safety endpoints.
Voclosporin for Focal Segmental Glomerulosclerosis (“FSGS”)
Aurinia initiated a Phase 2 proof-of-concept, open-label study for FSGS in June 2018. The study was designed to evaluate the role of voclosporin for treatment-naïve patients diagnosed with primary FSGS. In response to slower than anticipated enrollment, Aurinia has recently opened additional clinical trial sites outside of the United States and has amended the study to allow enrollment of FSGS patients who have previously received limited corticosteroid exposure. Up to approximately 20 patients are expected to be enrolled with interim results anticipated in 2020.
Financial Liquidity at June 30, 2019
As at June 30, 2019, Aurinia had cash and cash equivalents of $131.5 million compared to $144.3 million of cash, cash equivalents and short-term investments at March 31, 2019, and $125.9 million at December 31, 2018. Net cash used in operating activities was $13.3 million for the second quarter ended June 30, 2019, compared to $12.3 million for the second quarter ended June 30, 2018.
The Company believes, that based on its current plans that it has sufficient financial resources to fund the existing LN program, including the AURORA trial and the AURORA 2 extension trial, complete the NDA submission to the FDA, conduct the ongoing Phase 2 study for FSGS, initiate the AUDREY Phase 2/3 study, and fund operations into the second half of 2020.
Second Quarter 2019 Financial Results
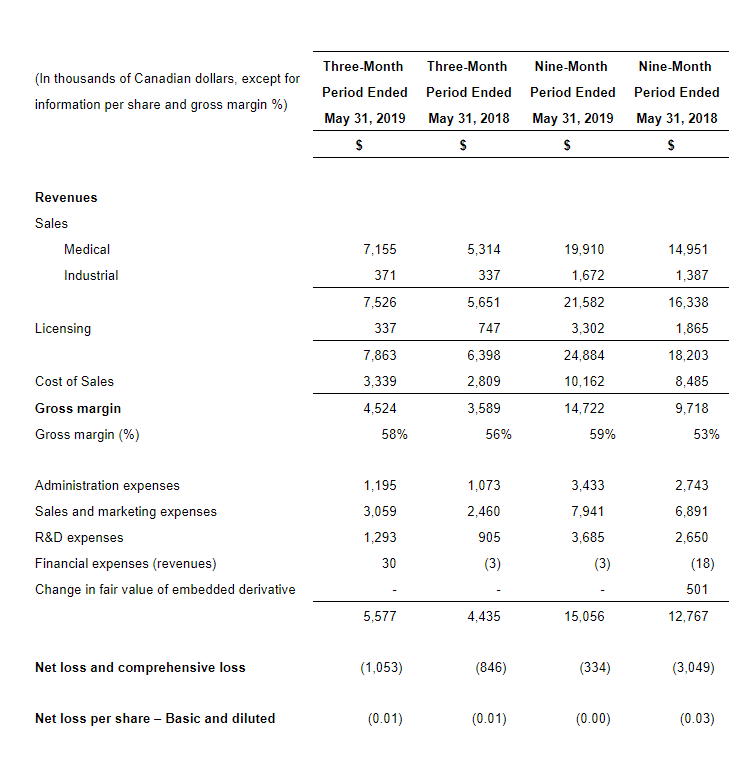
For the three months ended June 30, 2019, Aurinia reported a consolidated net loss of $15.9 million or $0.17 per common share compared to a consolidated net loss of $15.7 million or $0.19 per common share for the same period in 2018.
Research and development expenses (R&D) increased slightly to $11.2 million for the three months ended June 30, 2019, compared to $10.5 million for the three months ended June 30, 2018. The increase in these expenses reflected higher costs incurred for the AURORA 2 extension trial, the drug-drug interaction (“DDI”) study preparation costs associated with the planned NDA submission for LN and preparation costs for the Phase 2/3 DES clinical study.
Corporate, administration and business development expenses increased to $4.9 million for the three months ended June 30, 2019, compared to $3.5 million for the same period in 2018. The increase was primarily due to an increase in consulting fees related to recruitment fees and pre-commercial activities, such as market and payor research, and higher personnel and sponsorship costs.
Non-cash stock compensation expense was $2.0 million for the second quarter ended June 30, 2019, as compared with $2.0 million for the same period in 2018 and is included in both research and development and corporate, general and business development expenses.
Aurinia incurred other expenses of $720,000 during the three months ended June 30, 2019, associated with the successful defense of a proxy contest in connection with its June 26, 2019 annual general meeting. There was no similar expense in the comparable period.
Aurinia also recorded a non-cash reduction of $625,000 in the estimated fair value of derivative warrant liabilities which reduced the loss for the second quarter ended June 30, 2019 compared to an increase of $1.9 million in the estimated fair value of derivative warrant liabilities which increased the loss for the second quarter ended June 30, 2018. The derivative warrant liabilities will ultimately be eliminated on the exercise or forfeiture of the warrants and will not result in any cash outlay by the Company.
Financial Results for Six Months Ended June 30, 2019
For the six months ended June 30, 2019, Aurinia reported a consolidated net loss of $28.3 million or $0.31 per common share compared to a consolidated net loss of $31.2 million or $0.37 per common share for the comparable period in 2018.
R&D expenses were $21.8 million for the six months ended June 30, 2019, compared to $19.4 million for the same period in 2018. The increase in these expenses reflected higher costs incurred for the AURORA 2 extension trial, the DDI study and preparation costs associated with the planned LN NDA submission partially offset by lower AURORA trial costs as this trial nears completion.
Corporate, administration and business development expenses were $8.8 million for the six months ended June 30, 2019, compared to $7.3 million for the same period in 2018. The increase reflects the same items as noted in the second quarter corporate, administration and business development expenses.
Non-cash stock compensation expense totaled $3.6 million for the six months ended June 30, 2019, as compared with $4.1 million for the same period in 2018 and is included in both research and development and corporate, general and business development expenses.
For the six months ended June 30, 2019, Aurinia recorded a decrease of $2.4 million in the estimated fair value of derivative warrant liabilities compared to an increase of $4.6 million for the comparable period in 2018.
Unaudited interim condensed consolidated financial statements and the Management’s Discussion and Analysis are available on Aurinia’s website, SEDAR and EDGAR.
About Aurinia
Aurinia Pharmaceuticals is a late clinical-stage biopharmaceutical company focused on developing and commercializing therapies to treat targeted patient populations that are impacted by serious diseases with a high unmet medical need. The Company is currently developing an investigational drug, for the treatment of Lupus Nephritis, Focal Segmental Glomerulosclerosis, and Dry Eye Syndrome. The Company’s head office is in Victoria, British Columbia and focuses its development efforts globally.
About Voclosporin
Voclosporin, an investigational drug, is a novel and potentially best-in-class calcineurin inhibitor (“CNI”) with clinical data in over 2,600 patients across indications. Voclosporin is an immunosuppressant, with a synergistic and dual mechanism of action. By inhibiting calcineurin, voclosporin blocks IL-2 expression and T-cell mediated immune responses and stabilizes the podocyte in the kidney. It has been shown to have a more predictable pharmacokinetic and pharmacodynamic relationship (potentially requires no therapeutic drug monitoring), an increase in potency (vs cyclosporin), and an improved metabolic profile compared to legacy CNIs. Aurinia anticipates that upon regulatory approval, patent protection for voclosporin will be extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act and comparable laws in other countries and until April 2028 with anticipated pediatric extension. Further, the new Notice of Allowanceis expected to result in the issuance of a U.S. patent with a term extending to December 2037. If the FDA approves the use of voclosporin for LN and the label for such use follows the dosing protocol under the Notice of Allowance, the issuance of this patent will expand the scope of intellectual property protection for voclosporin to December 2037.
About VOS
Voclosporin ophthalmic solution (“VOS”) is an aqueous, preservative-free nanomicellar solution intended for use in the treatment of DES. A Phase 2a study was recently completed with results released in January of 2019. Previously, a Phase 1 study with healthy volunteers and patients with DES was also completed as were studies in rabbit and dog models. VOS has IP protection until 2031.
About LN
Lupus Nephritis (“LN”) in an inflammation of the kidney caused by Systemic Lupus Erythematosus (“SLE”) and represents a serious progression of SLE. SLE is a chronic, complex and often disabling disorder. The disease is highly heterogeneous, affecting a wide range of organs and tissue systems. Unlike SLE, LN has straightforward disease outcomes (measuring proteinuria) where an early response correlates with long-term outcomes. In patients with LN, renal damage results in proteinuria and/or hematuria and a decrease in renal function as evidenced by reduced estimated glomerular filtration rate (“eGFR”), and increased serum creatinine levels. LN is debilitating and costly and if poorly controlled, LN can lead to permanent and irreversible tissue damage within the kidney, resulting in end-stage renal disease (“ESRD”), thus making LN a serious and potentially life-threatening condition.
About FSGS
Focal segmental glomerulosclerosis (“FSGS”) is a rare disease that attacks the kidney’s filtering units (glomeruli) causing serious scarring which leads to permanent kidney damage and even renal failure. FSGS is one of the leading causes of Nephrotic Syndrome (“NS”) and is identified by biopsy and proteinuria. NS is a collection of signs and symptoms that indicate kidney damage, including large amounts of protein in the urine; low levels of albumin and higher than normal fat and cholesterol levels in the blood, and edema. Similar to LN, early clinical response (measured by reduction of proteinuria) is thought to be critical to long-term kidney health in patients with FSGS. Currently, there are no approved therapies for FSGS in the United States and the European Union.
About DES
Dry eye syndrome (“DES”) is characterized by irritation and inflammation that occurs when the eye’s tear film is compromised by reduced tear production, imbalanced tear composition, or excessive tear evaporation. The impact of DES ranges from subtle, yet constant eye irritation to significant inflammation and scarring of the eye’s surface. Discomfort and pain resulting from DES can reduce the quality of life and cause difficulty reading, driving, using computers and performing daily activities. DES is a chronic disease. There are currently three FDA approved therapies for the treatment of dry eye; however, there is an opportunity for potential improvement in the effectiveness by enhancing tolerability, the onset of action and alleviating the need for repetitive dosing.
Forward-Looking Statements
Certain statements made in this press release may constitute forward-looking information within the meaning of applicable Canadian securities law and forward-looking statements within the meaning of applicable United States securities law. These forward-looking statements or information include but are not limited to statements or information with respect to: AURORA having data around the end of this year, completing NDA submissions in a successful and timely manner including the anticipated NDA filing during the first half of next year and subsequent commercial launch in 2021, voclosporin being potentially a best-in-class CNI with robust intellectual property exclusivity; the anticipated AUDREY clinical study including enrolling the first subject in the fourth quarter of 2019 and the number of subjects expected to be enrolled; the expected timing of FSGS results and patient enrollment; and that Aurinia has sufficient financial resources to fund the existing LN program, including the AURORA trial, and the NDA submission to the FDA, conduct the current Phase 2a study for FSGS, commence additional studies for DES and fund operations into the second half of 2020. Aurinia’s anticipation that upon regulatory approval, patent protection for voclosporin will be extended in the United States and certain other major markets, including Europe and Japan, until at least October 2027 under the Hatch-Waxman Act and comparable laws in other countries and until April 2028 with anticipated pediatric extension; that the new Notice of Allowance is expected to result in the issuance of a U.S. patent with a term extending to December 2037; that if the FDA approves the use of voclosporin for LN and the label for such use follows the dosing protocol under the Notice of Allowance, the issuance of this patent will expand the scope of intellectual property protection for voclosporin to December 2037. It is possible that such results or conclusions may change based on further analyses of these data. Words such as “anticipate”, “will”, “believe”, “estimate”, “expect”, “intend”, “target”, “plan”, “goals”, “objectives”, “may” and other similar words and expressions, identify forward-looking statements. We have made numerous assumptions about the forward-looking statements and information contained herein, including among other things, assumptions about: the market value for the LN, DES and FSGS programs; that another company will not create a substantial competitive product for Aurinia’s LN, DES and FSGS business without violating Aurinia’s intellectual property rights; the burn rate of Aurinia’s cash for operations; the costs and expenses associated with Aurinia’s clinical trials; the planned studies achieving positive results; Aurinia being able to extend and protect its patents on terms acceptable to Aurinia; and the size of the LN, DES or FSGS markets. Even though the management of Aurinia believes that the assumptions made, and the expectations represented by such statements or information are reasonable, there can be no assurance that the forward-looking information will prove to be accurate.
Forward-looking information by their nature are based on assumptions and involve known and unknown risks, uncertainties and other factors which may cause the actual results, performance or achievements of Aurinia to be materially different from any future results, performance or achievements expressed or implied by such forward-looking information. Should one or more of these risks and uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those described in forward-looking statements or information. Such risks, uncertainties and other factors include, among others, the following: difficulties, delays, or failures we may experience in the conduct of our clinical trial; difficulties we may experience in completing the development and commercialization of voclosporin; the market for the LN, DES and FSGS business may not be as estimated; Aurinia may have to pay unanticipated expenses; estimated costs for clinical trials may be underestimated, resulting in Aurinia having to make additional expenditures to achieve its current goals; Aurinia not being able to extend or fully protect its patent portfolio for voclosporin; and competitors may arise with similar products. Although we have attempted to identify factors that could cause actual actions, events or results to differ materially from those described in forward-looking statements and information, there may be other factors that cause actual results, performances, achievements or events to not be as anticipated, estimated or intended. Also, many of the factors are beyond our control. There can be no assurance that forward-looking statements or information will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, you should not place undue reliance on forward-looking statements or information.
Except as required by law, Aurinia will not update forward-looking information. All forward-looking information contained in this press release is qualified by this cautionary statement. Additional information related to Aurinia, including a detailed list of the risks and uncertainties affecting Aurinia and its business can be found in Aurinia’s most recent Annual Information Form available by accessing the Canadian Securities Administrators’ System for Electronic Document Analysis and Retrieval (SEDAR) website at www.sedar.com or the U.S. Securities and Exchange Commission’s Electronic Document Gathering and Retrieval System (EDGAR) website at www.sec.gov/edgar.