SOUTH SAN FRANCISCO, Calif. (GLOBE NEWSWIRE) — Satsuma Pharmaceuticals, Inc. (Nasdaq: STSA), a clinical-stage biopharmaceutical company, today reported financial results for the quarter and full-year ended December 31, 2019 and summarized recent business results.
Related Article: Satsuma Pharmaceuticals Appoints Rob Janosky as Chief Commercial Officer
“In 2019, the Satsuma team made significant progress toward its goal of delivering STS101, a compact, simple-to-use, self-administered, and non-injectable DHE (or dihydroergotamine) product, as a differentiated treatment option for people with migraine. We’ve established a strong foundation for our business and in 2020 we plan to advance Phase 3 development of STS101 and begin preparing for an NDA filing by the end of 2021 and subsequent STS101 commercial introduction.”
– John Kollins, Satsuma’s President & CEO
Financial Results for Q4 2019 & FY2019
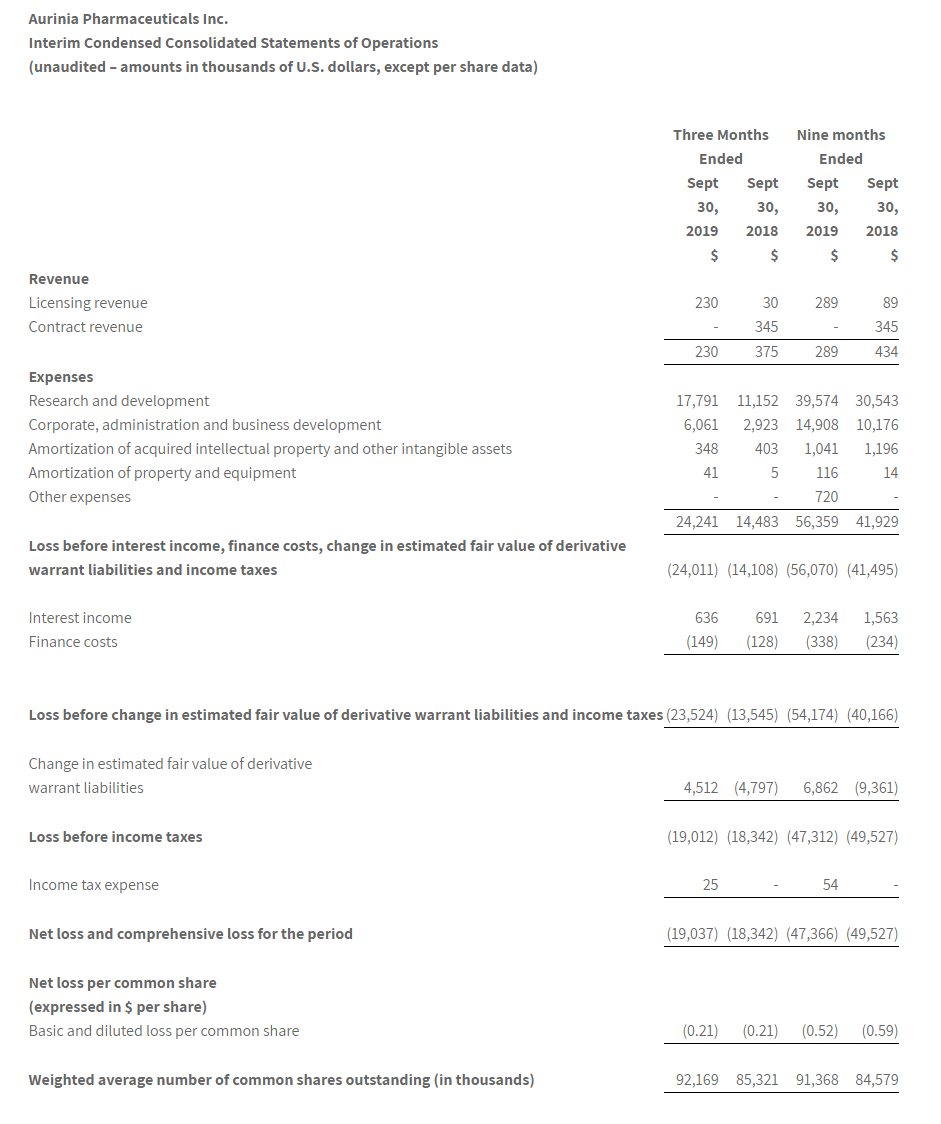
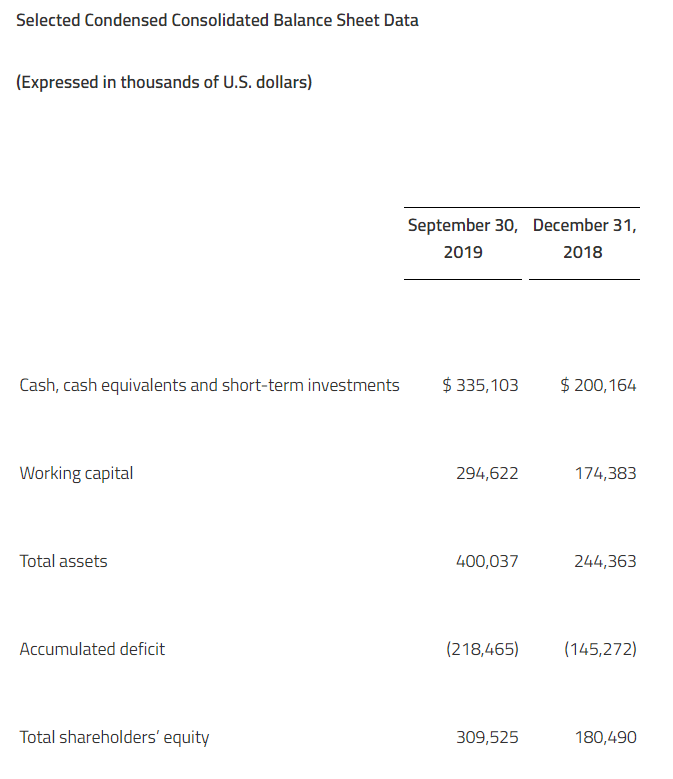
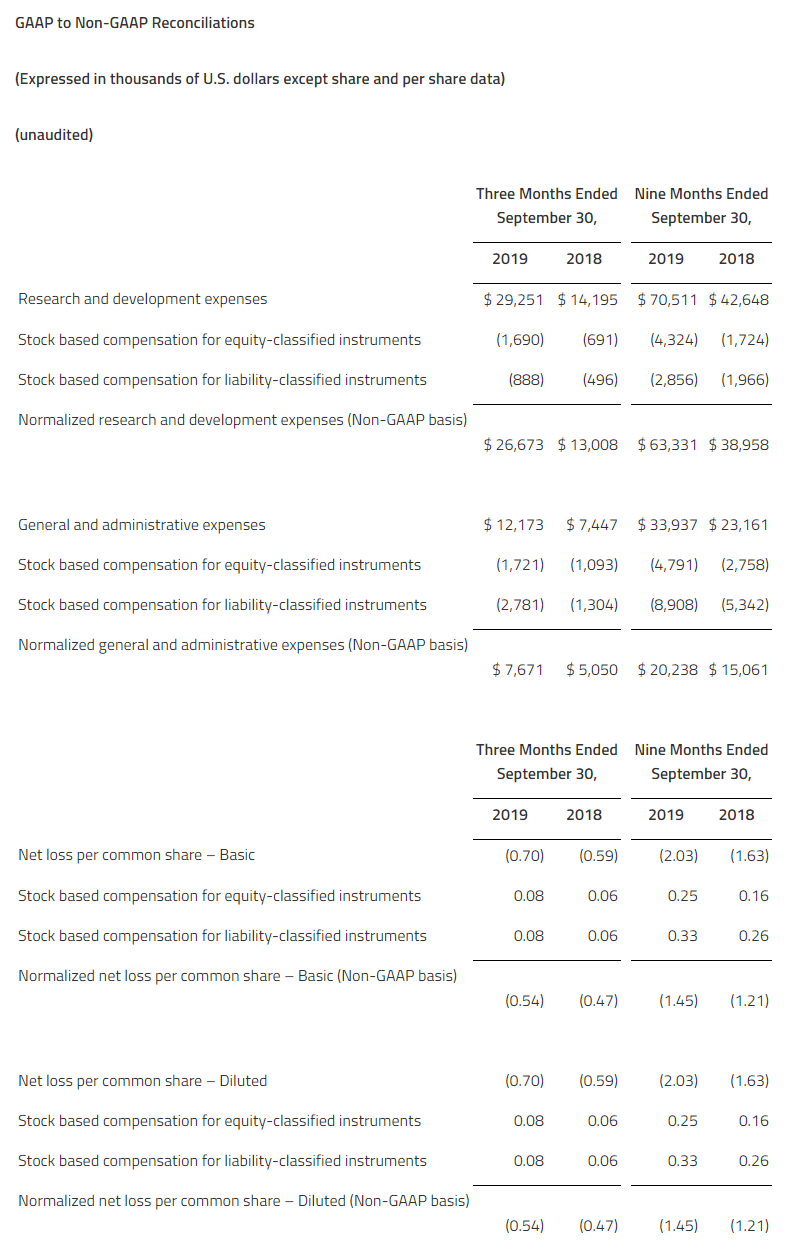
Net losses for the fourth quarter and full-year 2019 were $10.8 million and $28.2 million, respectively, or $0.62 and $4.80 per common share, respectively. This compared to net losses of $2.3 million and $7.3 million, respectively, or $2.17 and $7.15 per common share, respectively for the same periods in 2018. As of December 31, 2019, the Company had $117.9 million of cash, cash equivalents, and marketable securities. The Company believes it has sufficient financial resources to fund operations through the end of 2021.
Research and development expenses were $9.2 million and $24.2 million for the fourth quarter and full-year 2019, respectively, compared to $2.0 million and $6.4 million for the same periods of 2018, respectively. Fourth-quarter expenses increased by $7.2 million, primarily due to additional expenses for the EMERGE clinical trial and drug supply manufacturing activities, as well as increases in salaries and employee-related expenses.
General and administrative expenses were $2.1 million and $4.7 million for the fourth quarter and full-year 2019, respectively, compared to $0.3 million and $1.1 million for the same periods of 2018, respectively. Fourth-quarter expenses increased by $1.8 million, primarily due to general administrative expenses, as well as increases in salaries and employee-related expenses.
Business Update & FY 2019 Highlights
STS101 EMERGE Phase 3 Efficacy Trial Update
Satsuma expects to report topline data for the EMERGE trial in the second half of 2020. The EMERGE trial, which Satsuma believes is the largest-ever clinical trial undertaken with any DHE product, is a double-blind, parallel-group, placebo-controlled, multicenter trial in approximately 1,140 patients. EMERGE is designed to evaluate the efficacy, safety, and tolerability of STS101 in treating a single migraine attack and is highly powered on its two co-primary endpoints: freedom from pain (>99% power) and freedom from most-bothersome-symptom (from among photophobia, phonophobia or nausea) (>95% power) at two hours after administration of study medication. In addition, EMERGE is designed to prospectively evaluate a number of secondary endpoints and the performance of STS101 in patient subgroups that could enhance its differentiated clinical profile.
The EMERGE trial is the first of two Phase 3 trials Satsuma plans to complete to support registration of STS101. Satsuma plans to initiate in the third quarter of 2020 an open-label, Phase 3 safety trial of STS101 in at least 150 episodic migraine patients, which will treat their migraines with STS101 on an as-needed basis for six months, with a possible 50 patients treated for an additional six months.
STS101 Phase 1 clinical trial results presented in July at the American Headache Society’s Annual Scientific Meeting and subsequently published in its official peer-reviewed journal, HEADACHE: The Journal of Head and Face Pain
The STS101 Phase 1 trial was an open-label, two-part, active-controlled, three-period crossover study designed to investigate and compare the safety and pharmacokinetics of STS101, DHE liquid nasal spray (Migranal®), and intramuscular (IM) DHE injection in healthy subjects.
Study authors concluded that STS101 showed a favorable tolerability profile and was rapidly absorbed, achieving DHE plasma concentrations comparable to IM DHE and exceeding Migranal. Based on data from this study and results from other clinical studies with DHE (including injected, liquid nasal spray, and orally inhaled DHE dosage forms), the authors posited that STS101 is anticipated to demonstrate rapid pain relief, improvement in patient function, and excellent 2-hour and sustained pain freedom rates.
Two STS101 posters presented by Satsuma at the 19th Congress of the International Headache Society comparing the PK of STS101 with other DHE formulations and demonstrating the consistent and robust delivery performance of STS101
These posters are available for download on the Publications section of the Satsuma Pharmaceuticals website.
Financing activities and corporate update
2019 financing activities raised approximately $153 million:
- In April 2019, Satsuma closed a $62 million Series B preferred financing.
- In September and October 2019, Satsuma raised nearly $91 million in aggregate gross proceeds in its initial public offering, which resulted in the Company’s shares being listed on the Nasdaq Global Market.
Expanded leadership team, bolstering financial and commercial management functions and corporate governance:
- In May 2019, Satsuma appointed Elisabeth Sandoval to its Board of Directors. Ms. Sandoval brings to Satsuma extensive drug product commercialization experience, as well as leadership and strategic skills in the migraine field. Ms. Sandoval most recently served as Chief Commercial Officer and Executive Vice President of Corporate Strategy for Alder Biopharmaceuticals (acquired in 2019 by Lundbeck), which recently received FDA approval for eptinezumab, a novel therapeutic antibody for the preventive treatment of migraine, and previously served as Chief Commercial Officer of KYTHERA Biopharmaceuticals.
- In February 2019, Tom O’Neil joined Satsuma as its Chief Financial Officer. Prior to joining Satsuma Pharmaceuticals, Mr. O’Neil was Chief Financial Officer at Protagonist Therapeutics, Inc. Since entering the biopharmaceutical industry in 1999, Mr. O’Neil has served in C-level and other management roles in multiple public and private biopharmaceutical companies. He brings to Satsuma a wealth of relevant finance and operations experience, including leading successful initial public offering and private equity transaction initiatives.
- In March 2020, Rob Janosky joined the Company as Chief Commercial Officer. Mr. Janosky’s experience in the biopharmaceutical industry spans more than 25 years and includes successfully creating partnerships, building commercial capabilities and launching products within established and emerging biopharmaceutical companies, including DURECT Corporation, Jazz Pharmaceuticals, Vivus Inc., Johnson & Johnson/Alza Corporation, and Wyeth.
Upcoming 2020 Milestones
- EMERGE Phase 3 efficacy trial top-line data in the second half of 2020
- Initiate STS101 Phase 3 open-label safety trial in the third quarter of 2020
- Present further data on STS101, DHE, and the proprietary dry-powder nasal drug delivery technologies incorporated in STS101 at medical meetings in 2020
About Satsuma Pharmaceuticals and STS101
Satsuma Pharmaceuticals is a clinical-stage biopharmaceutical company developing a novel therapeutic product for the acute treatment of migraine, STS101. STS101 is a drug-device combination of a proprietary dry-powder formulation of dihydroergotamine mesylate (DHE), which can be quickly and easily self-administered with a proprietary pre-filled, single-use, nasal delivery device. In developing STS101, Satsuma has applied proprietary nasal drug delivery, dry-powder formulation, and engineered drug particle technologies to create a compact, simple-to-use, non-injectable DHE product that can be rapidly self-administered in a matter of seconds. The Company believes STS101 would, if approved, be an attractive migraine treatment option for many patients and may enable a larger number of people with migraine to realize the long-recognized therapeutic benefits of DHE therapy. STS101 has undergone extensive pre-clinical development, completed a Phase 1 clinical trial, and is currently in Phase 3 development.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements concerning the business, operations and financial performance and condition of Satsuma Pharmaceuticals, Inc. (the “Company”), as well as the Company’s plans, objectives and expectations for its business operations and financial performance and condition. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “aim,” “anticipate,” “assume,” “believe,” “contemplate,” “continue,” “could,” “due,” “estimate,” “expect,” “goal,” “intend,” “may,” “objective,” “plan,” “predict,” “potential,” “positioned,” “seek,” “should,” “target,” “will,” “would,” and other similar expressions that are predictions of or indicate future events and future trends, or the negative of these terms or other comparable terminology. These forward-looking statements include, but are not limited to, statements about the Company’s expectations regarding the potential safety and efficacy of STS101; the Company’s clinical and regulatory development plans; the Company’s expectations with regard to the initiation and availability of data to be derived from its ongoing and planned Phase 3 clinical trials; the timing and likelihood of regulatory filings and approvals for STS101; and expected cash needs and sufficiency of cash on hand. In light of these risks and uncertainties, the events or circumstances referred to in the forward-looking statements may not occur. The Company’s actual results could differ materially from those stated or implied in forward-looking statements due to a number of factors, including but not limited to, risks detailed in the Company’s Annual Report on Form 10-K for the year ended December 31, 2019, filed with the Securities and Exchange Commission, as well as other documents that may be filed by the Company from time to time. In particular, the following factors, among others, could cause results to differ materially from those expressed or implied by such forward-looking statements: the Company’s ability to demonstrate sufficient evidence of efficacy and safety in its clinical trials of STS101; the results of preclinical and clinical studies may not be predictive of future results; the unpredictability of the regulatory process; regulatory developments in the United States and foreign countries; the costs of clinical trials may exceed expectations; and the Company’s ability to raise additional capital. Although the Company believes that the expectations reflected in the forward-looking statements are reasonable, it cannot guarantee that the events and circumstances reflected in the forward-looking statements will be achieved or occur, and the timing of events and circumstances and actual results could differ materially from those projected in the forward-looking statements. Accordingly, you should not place undue reliance on these forward-looking statements. All such statements speak only as of the date made, and the Company undertakes no obligation to update or revise publicly any forward-looking statements, whether as a result of new information, future events or otherwise.
This press release discusses STS101, a product candidate that is in clinical development, and has not yet been approved for marketing by the U.S. Food and Drug Administration. No representation is made as to the safety or effectiveness of STS101 for the therapeutic use for which STS101 is being studied.
INVESTOR AND CORPORATE CONTACTS:
Corey Davis, PhD
LifeSci Advisors, LLC
cdavis@lifesciadvisors.com
Tom O’Neil, Chief Financial Officer
Satsuma Pharmaceuticals, Inc.
tom@satsumarx.com